

Impact of Pre-Extraction Methods on Apple Blossom Microbiome Analysis
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Orchards, Treatments, and Sampling
2.2. Processing of Blossoms
2.2.1. Lyophilization
2.2.2. Grinding
2.2.3. Sonication
2.3. DNA Sequencing and Bioinformatics
2.4. Data Analysis
3. Results
3.1. Impact of Pre-Extraction Methods and PNA Use on Read Recovery
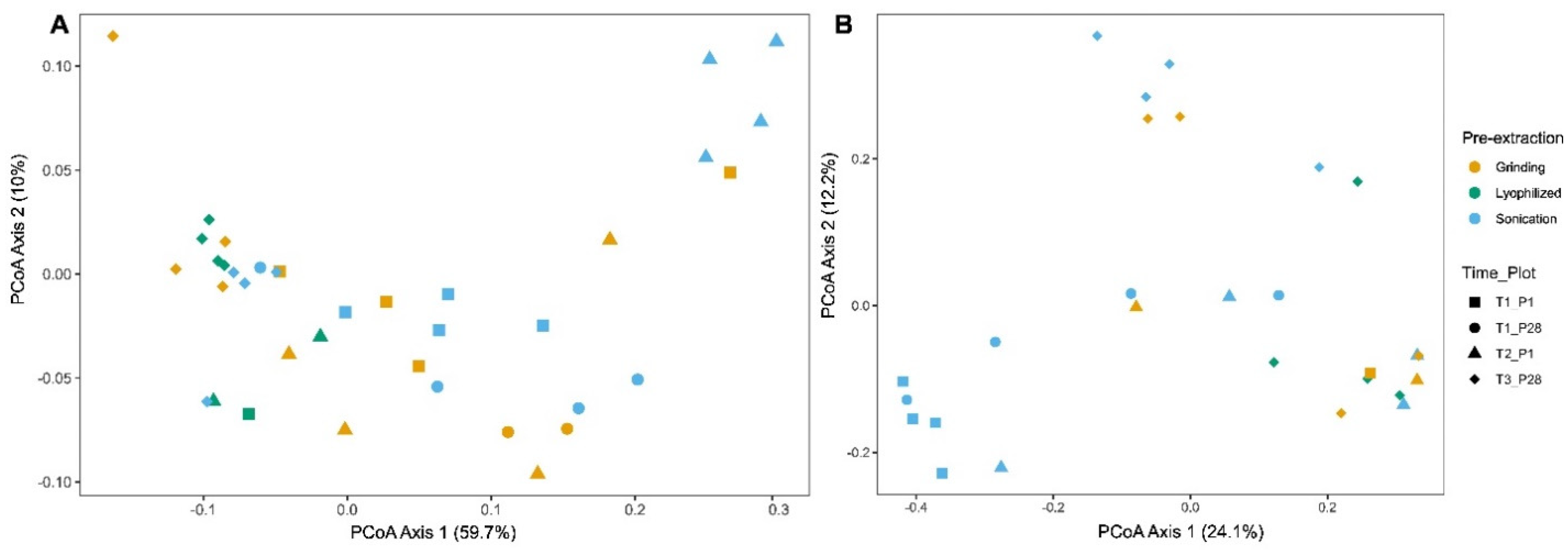
3.2. Effect of Pre-Extraction Methods on Alpha and Beta Diversity Across Time Points
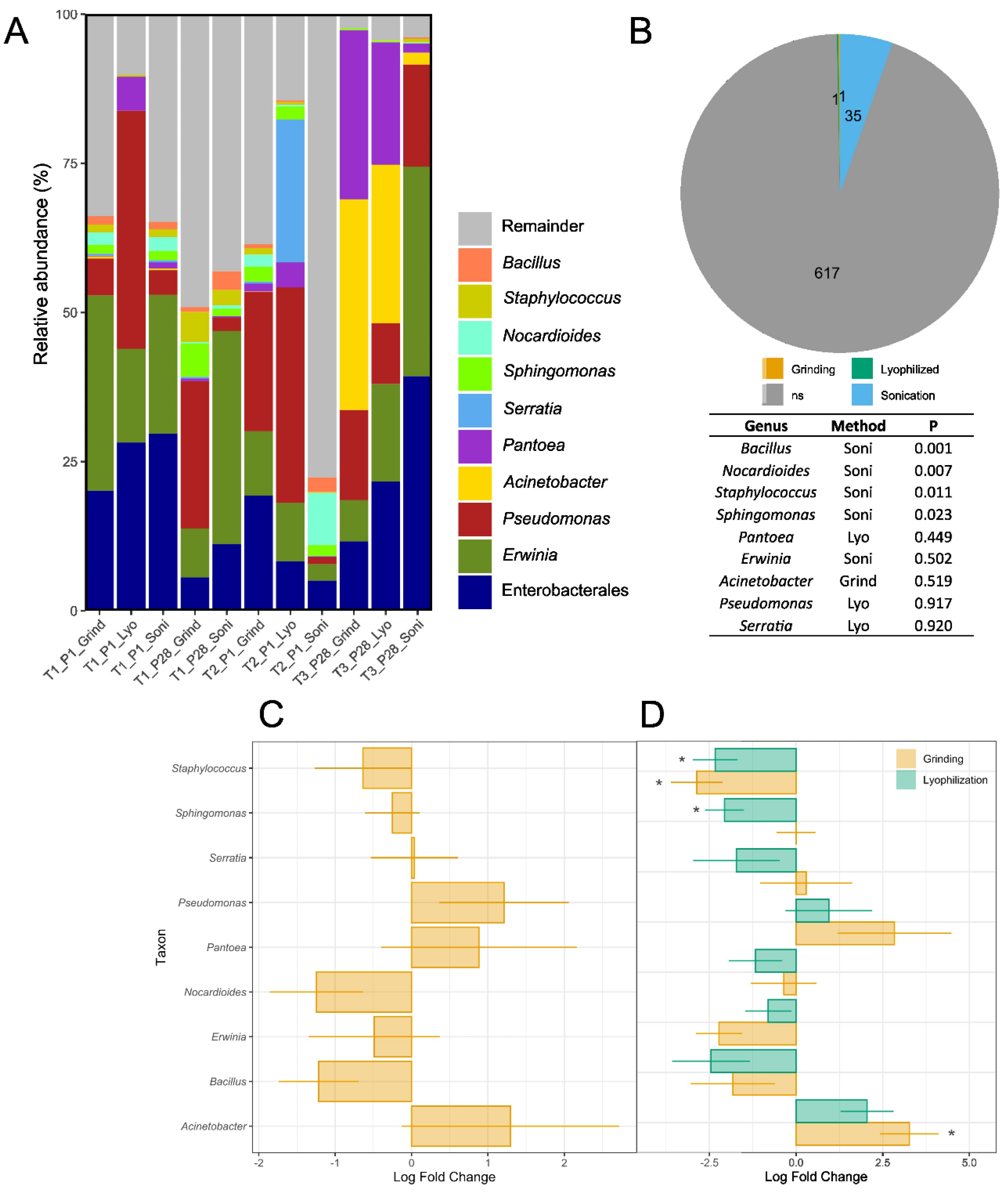
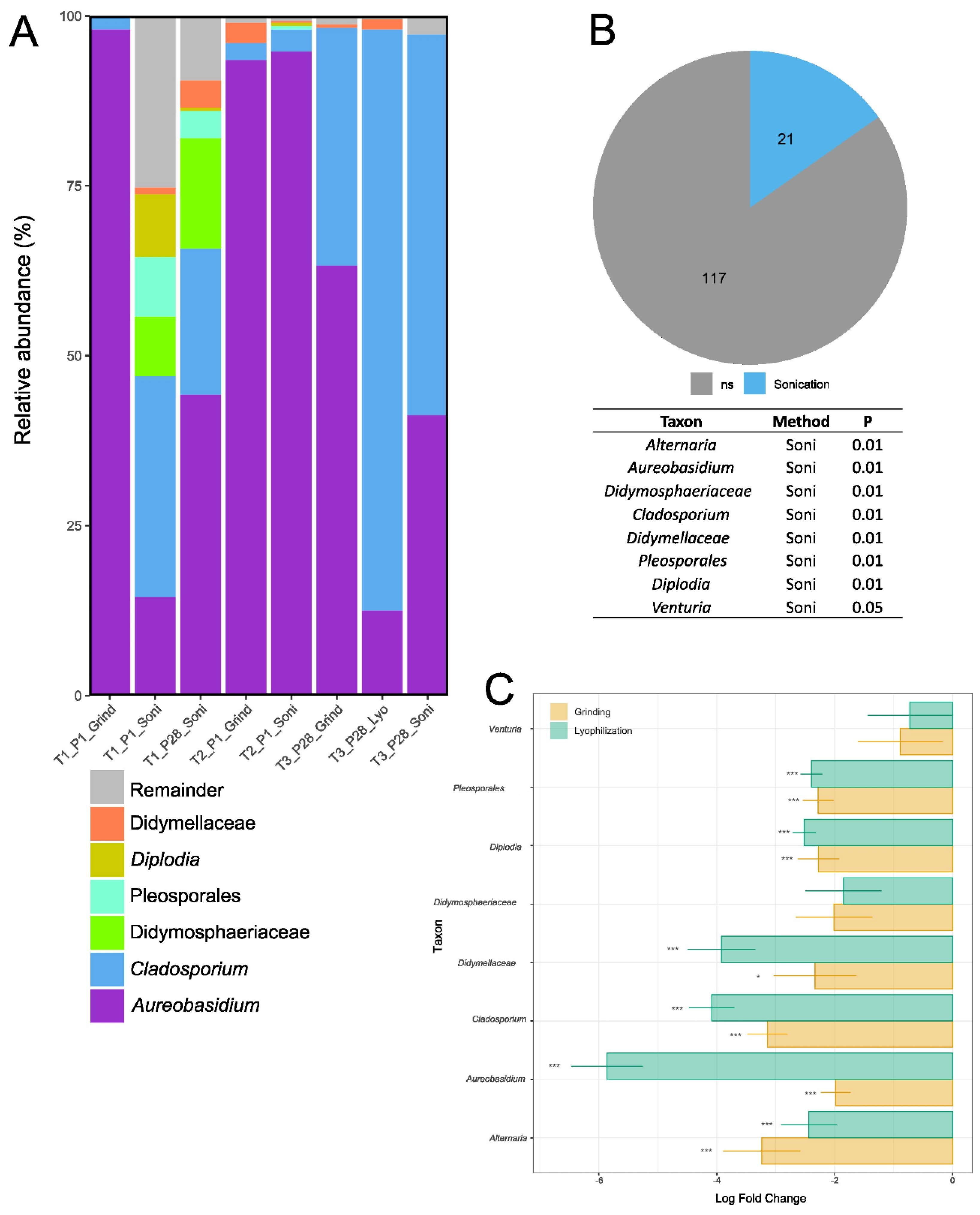
3.3. Taxonomic Analyses and Differential Abundance Testing of Bacterial and Fungal Genera
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vannette, R.L. The floral microbiome: Plant, pollinator, and microbial perspectives. Annu. Rev. Ecol. Evol. Syst. 2020, 51, 363–386. [Google Scholar] [CrossRef]
- Shade, A.; McManus, P.S.; Handelsman, J. Unexpected diversity during community succession in the apple flower microbiome. mBio 2013, 4, e00602-12. [Google Scholar] [CrossRef]
- Aleklett, K.; Hart, M.; Shade, A. The microbial ecology of flowers: An emerging frontier in phyllosphere research. Botany 2014, 92, 253–266. [Google Scholar] [CrossRef]
- Thomson, S. The role of the stigma in fire blight infections. Phytopathology 1986, 76, 476–482. [Google Scholar] [CrossRef]
- Cui, Z.; Huntley, R.B.; Zeng, Q.; Steven, B. Temporal and spatial dynamics in the apple flower microbiome in the presence of the phytopathogen Erwinia amylovora. ISME J. 2021, 15, 318–329. [Google Scholar] [CrossRef]
- Cui, Z.; Huntley, R.B.; Schultes, N.P.; Zeng, Q.; Steven, B. Inoculation of stigma-colonizing microbes to apple stigmas alters microbiome structure and reduces the occurrence of fire blight disease. Phytobiomes J. 2021, 5, 156–165. [Google Scholar] [CrossRef]
- Oz, A.; Mairesse, O.; Raikin, S.; Hanani, H.; Dafny Yelin, M.; Sharon, I. Pear flower and leaf microbiome dynamics during the naturally occurring spread of Erwinia amylovora. bioRxiv 2024. [Google Scholar] [CrossRef]
- Johnson, K.B.; Stockwell, V.O. Management of fire blight: A case study in microbial ecology. Annu. Rev. Phytopathol. 1998, 36, 227–248. [Google Scholar] [CrossRef]
- Thomson, S.V. Epidemiology of fire blight. In Fire Blight: The Disease and Its Causative Agent, Erwinia amylovora; Vanneste, J.L., Ed.; CABI Pub.: Oxon, UK, 2000; pp. 9–36. [Google Scholar]
- Gayder, S.; Kammerecker, S.; Fieseler, L. Biological control of the fire blight pathogen Erwinia amylovora using bacteriophages. J. Plant Pathol. 2023, 106, 853–869. [Google Scholar] [CrossRef]
- Van Der Zwet, T. Fire Blight—Its Nature, Prevention, and Control: A Practice Guide to Integrated Disease Management; US Department of Agriculture, Ithaca Cornell University Press: Ithaca, NY, USA, 1992; Volume 631, p. 83. [Google Scholar]
- Bubán, T.; Orosz-Kovács, Z. The nectary as the primary site of infection by Erwinia amylovora (Burr.) Winslow et al.: A mini review. Plant Syst. Evol. 2003, 238, 183–194. [Google Scholar] [CrossRef]
- Peil, A.; Bus, V.G.; Geider, K.; Richter, K.; Flachowsky, H.; Hanke, M.-V. Improvement of fire blight resistance in apple and pear. Int. J. Plant Breed 2009, 3, 1–27. [Google Scholar]
- Zeng, Q.; Johnson, K.B.; Mukhtar, S.; Nason, S.; Huntley, R.; Millet, F.; Yang, C.-H.; Hassani, M.A.; Zuverza-Mena, N.; Sundin, G.W. Aureobasidium pullulans from the fire blight biocontrol product, Blossom protect, induces host resistance in apple flowers. Phytopathology 2023, 113, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Steven, B.; Huntley, R.B.; Zeng, Q. The influence of flower anatomy and apple cultivar on the apple flower phytobiome. Phytobiomes J. 2018, 2, 171–179. [Google Scholar] [CrossRef]
- Pedroncelli, A.; Puopolo, G. This tree is on fire: A review on the ecology of Erwinia amylovora, the causal agent of fire blight disease. J. Plant Pathol. 2023, 106, 823–837. [Google Scholar] [CrossRef]
- Pusey, P.L.; Stockwell, V.O.; Rudell, D.R. Antibiosis and acidification by Pantoea agglomerans strain E325 may contribute to suppression of Erwinia amylovora. Phytopathology 2008, 98, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Puławska, J.; Schachterle, J. Early events in fire blight infection and pathogenesis of Erwinia amylovora. J. Plant Pathol. 2021, 103, 13–24. [Google Scholar] [CrossRef]
- Kharadi, R.R.; Sundin, G.W. Dissecting the process of xylem colonization through biofilm formation in Erwinia amylovora. J. Plant Pathol. 2021, 103, 41–49. [Google Scholar] [CrossRef]
- Sare, A.R.; Stouvenakers, G.; Eck, M.; Lampens, A.; Goormachtig, S.; Jijakli, M.H.; Massart, S. Standardization of plant microbiome studies: Which proportion of the microbiota is really harvested? Microorganisms 2020, 8, 342. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Lu-Irving, P.; Copeland, J.; Guttman, D.S.; Wang, P.W.; Baltrus, D.A.; Dlugosch, K.M.; Johnson, M.T.J. Chloroplast sequence variation and the efficacy of peptide nucleic acids for blocking host amplification in plant microbiome studies. Microbiome 2018, 6, 144. [Google Scholar] [CrossRef]
- Viquez, R.L.; Fleischer, R.; Wilhelm, K.; Tschapka, M.; Sommer, S. Jumping the green wall: The use of PNA-DNA clamps to enhance microbiome sampling depth in wildlife microbiome research. Ecol. Evol. 2020, 10, 11779–11786. [Google Scholar] [CrossRef]
- Borodušķe, A.; Ķibilds, J.; Fridmanis, D.; Gudrā, D.; Ustinova, M.; Seņkovs, M.; Nikolajeva, V. Does peptide-nucleic acid (PNA) clamping of host plant DNA benefit ITS1 amplicon-based characterization of the fungal endophyte community? Fungal. Ecol. 2023, 61, 101181. [Google Scholar] [CrossRef]
- Kawasaki, A.; Ryan, P.R. Peptide nucleic acid (PNA) clamps to reduce co-amplification of plant DNA during PCR amplification of 16S rRNA genes from endophytic bacteria. Methods Mol. Biol. 2021, 2232, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, E.; Gardner, C.M.; Gunsch, C.K. A novel PCR-clamping assay reducing plant host DNA amplification significantly improves prokaryotic endo-microbiome community characterization. FEMS Microbiol. Ecol. 2020, 96, fiaa110. [Google Scholar] [CrossRef]
- Lee, S.I.; Cho, G.; Kim, S.H.; Kim, D.R.; Kwak, Y.S. Mycobiota community and fungal species response to development stage and fire blight disease in apples. AIMS Microbiol. 2023, 9, 554–569. [Google Scholar] [CrossRef] [PubMed]
- Gdanetz, K.; Dobbins, M.; Villani, S.; Outwater, C.; Slack, S.; Nesbitt, D.; Svircev, A.; Lauwers, E.; Zeng, Q.; Cox, K.D.; et al. Multisite field evaluation of bacteriophages for fire blight management: Incorporation of UVR protectants, and impact on the apple flower microbiome. Phytopathology 2023, 114, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Slack, S.M.; Schachterle, J.K.; Sweeney, E.M.; Kharadi, R.R.; Peng, J.; Botti-Marino, M.; Bardaji, L.; Pochubay, E.A.; Sundin, G.W. In-orchard population dynamics of Erwinia amylovora on apple flower stigmas. Phytopathology 2022, 112, 1214–1225. [Google Scholar] [CrossRef]
- Schnyder, A.; Eberl, L.; Agnoli, K. Investigating the biocontrol potential of the natural microbiota of the apple blossom. Microorganisms 2022, 10, 2480. [Google Scholar] [CrossRef]
- Slack, S.M.; Walters, K.J.; Outwater, C.A.; Sundin, G.W. Effect of kasugamycin, oxytetracycline, and streptomycin on in-orchard population dynamics of Erwinia amylovora on apple flower stigmas. Plant Dis. 2021, 105, 1843–1850. [Google Scholar] [CrossRef]
- Kong, H.G.; Ham, H.; Lee, M.-H.; Park, D.S.; Lee, Y.H. Microbial community dysbiosis and functional gene content changes in apple flowers due to fire blight. Plant Pathol. J. 2021, 37, 404. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Freilich, S.; Bartuv, R.; Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Burchard, E.; Dardick, C.; et al. Global analysis of the apple fruit microbiome: Are all apples the same? Environ. Microbiol. 2021, 23, 6038–6055. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, C.; Ma, J.; Peng, Q.; Du, X.; Sun, S.e.; Cheng, J.e.; Peng, W.; Chen, L.; Gu, Z. Extraction methods determine the quality of soil microbiota acquisition. Microorganisms 2024, 12, 403. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, M.M.A.; Irfan, S.; Lorenzo, J.M.; Shafique, B.; Kanwal, R.; Pateiro, M.; Arshad, R.N.; Wang, L.; Nayik, G.A.; Roobab, U. Sonication, a potential technique for extraction of phytoconstituents: A systematic review. Processes 2021, 9, 1406. [Google Scholar] [CrossRef]
- Starke, R.; Jehmlich, N.; Alfaro, T.; Dohnalkova, A.; Capek, P.; Bell, S.L.; Hofmockel, K.S. Incomplete cell disruption of resistant microbes. Sci. Rep. 2019, 9, 5618. [Google Scholar] [CrossRef]
- Lindow, S.E. Epiphytic growth and survival. In Methods in Microbiology; Williams, P., Ketley, J., Salmond, G., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 27, pp. 177–182. [Google Scholar]
- McNees, C.R.; Law, A.D.; Moe, L.A. Characterization of endophytic microbial communities in store-bought kale evaluated by different plant tissue homogenization methods. Phytobiomes J. 2020, 4, 211–216. [Google Scholar] [CrossRef]
- Heckly, R.J. Preservation of bacteria by lyophilization. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 1961; Volume 3, pp. 1–76. [Google Scholar]
- Zhang, B.; Brock, M.; Arana, C.; Dende, C.; van Oers, N.S.; Hooper, L.V.; Raj, P. Impact of bead-beating intensity on the genus-and species-level characterization of the gut microbiome using amplicon and complete 16S rRNA gene sequencing. Front. Cell. Infect. Microbiol. 2021, 11, 678522. [Google Scholar] [CrossRef]
- Avramia, I.; Amariei, S. A simple and efficient mechanical cell disruption method using glass beads to extract β-glucans from spent brewer’s yeast. Appl. Sci. 2022, 12, 648. [Google Scholar] [CrossRef]
- Cullen, J.T.; Lawlor, P.G.; Cormican, P.; Crispie, F.; Gardiner, G.E. Optimisation of a bead-beating procedure for simultaneous extraction of bacterial and fungal DNA from pig faeces and liquid feed for 16S and ITS2 rDNA amplicon sequencing. Anim. Open Space 2022, 1, 100012. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef]
- Op De Beeck, M.; Lievens, B.; Busschaert, P.; Declerck, S.; Vangronsveld, J.; Colpaert, J.V. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS ONE 2014, 9, e97629. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rivers, A.R.; Weber, K.C.; Gardner, T.G.; Liu, S.; Armstrong, S.D. ITSxpress: Software to rapidly trim internally transcribed spacer sequences with quality scores for marker gene analysis. F1000Research 2018, 7, 1418. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Bok, G.; Ryberg, M.; Kristiansson, E.; Hallenberg, N. A software pipeline for processing and identification of fungal ITS sequences. Source Code Biol. Med. 2009, 4, 1–6. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: Sequences, taxa and classifications reconsidered. Nucleic Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Lozupone, C.; Hamady, M.; Knight, R. UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform. 2006, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Simpson, G.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Solymos, P.; Stevens, M.; Szoecs, E.; et al. vegan: Community Ecology Package. R Package Version 2.6-10. 2025. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 April 2024).
- Cáceres, M.D.; Legendre, P. Associations between species and groups of sites: Indices and statistical inference. Ecology 2009, 90, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]
- Taerum, S.J.; Steven, B.; Gage, D.J.; Triplett, L.R. Validation of a PNA clamping method for reducing host DNA amplification and increasing eukaryotic diversity in rhizosphere microbiome studies. Phytobiomes J. 2020, 4, 291–302. [Google Scholar] [CrossRef]
- Rezadoost, M.H.; Kordrostami, M.; Kumleh, H.H. An efficient protocol for isolation of inhibitor-free nucleic acids even from recalcitrant plants. 3 Biotech 2016, 6, 61. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Boutin, S.; Lussier, E.; Laforest-Lapointe, I. Investigating the spatiotemporal dynamics of apple tree phyllosphere bacterial and fungal communities across cultivars in orchards. Can. J. Microbiol. 2024, 70, 238–251. [Google Scholar] [CrossRef]
- Temple, T.N.; Thompson, E.C.; Uppala, S.; Granatstein, D.; Johnson, K.B. Floral colonization dynamics and specificity of Aureobasidium pullulans strains used to suppress fire blight of pome fruit. Plant Dis. 2020, 104, 121–128. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial 16S rRNA Reads Recovered After Rarefying and Filtering (%) | Fungal ITS Reads Recovered After Rarefying and Filtering (%) | ||||||
|---|---|---|---|---|---|---|---|
| Method | Orchard | T1 | T2 | T3 | T1 | T2 | T3 |
| Grinding | Plot 1 | 3.500 ± 2.200 (4/4) | 4.600 ± 1.500 (4/4) | − | 0.053 ± 0.100 (1/4) | 0.055 ± 0.050 (2/4) | − |
| Plot 28 | 0.900 ± 0.400 (4/4) | − | 39.900 ± 15.300 (4/4) | 0.009 ± 0.007 (0/4) | − | 1.210 ± 0.520 (4/4) | |
| Lyophilization | Plot 1 | 0.500 ± 0.500 (1/4) | 1.400 ± 2.900 (2/4) | − | 0.003 ± 0.005 (0/4) | 0.003 ± 0.003 (0/4) | − |
| Plot 28 | 0.300 ± 0.100 (0/4) | − | 21.900 ± 18.700 (4/4) | 0.000 ± 0.000 (0/4) | − | 0.190 ± 0.070 (4/4) | |
| Sonication | Plot 1 | 7.700 ± 6.800 (4/4) | 8.200 ± 5.900 (4/4) | − | 0.320 ± 0.240 (4/4) | 5.300 ± 2.000 (4/4) | − |
| Plot 28 | 7.100 ± 5.300 (4/4) | − | 95.700 ± 5.700 (4/4) | 0.670 ± 0.710 (4/4) | − | 45.800 ± 8.700 (4/4) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, N.N.; Gaiero, J.R.; Sulman, M.; Moote, P.; Nesbitt, D.; Svircev, A.M.; Ellouze, W. Impact of Pre-Extraction Methods on Apple Blossom Microbiome Analysis. Microorganisms 2025, 13, 923. https://doi.org/10.3390/microorganisms13040923
Patel NN, Gaiero JR, Sulman M, Moote P, Nesbitt D, Svircev AM, Ellouze W. Impact of Pre-Extraction Methods on Apple Blossom Microbiome Analysis. Microorganisms. 2025; 13(4):923. https://doi.org/10.3390/microorganisms13040923
Chicago/Turabian StylePatel, Nikhil N., Jonathan R. Gaiero, Muhammad Sulman, Paul Moote, Darlene Nesbitt, Antonet M. Svircev, and Walid Ellouze. 2025. "Impact of Pre-Extraction Methods on Apple Blossom Microbiome Analysis" Microorganisms 13, no. 4: 923. https://doi.org/10.3390/microorganisms13040923
APA StylePatel, N. N., Gaiero, J. R., Sulman, M., Moote, P., Nesbitt, D., Svircev, A. M., & Ellouze, W. (2025). Impact of Pre-Extraction Methods on Apple Blossom Microbiome Analysis. Microorganisms, 13(4), 923. https://doi.org/10.3390/microorganisms13040923