
The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5

Abstract
:
1. The Microbiome and Human Health
2. Role of the Microbiome in BA Metabolism
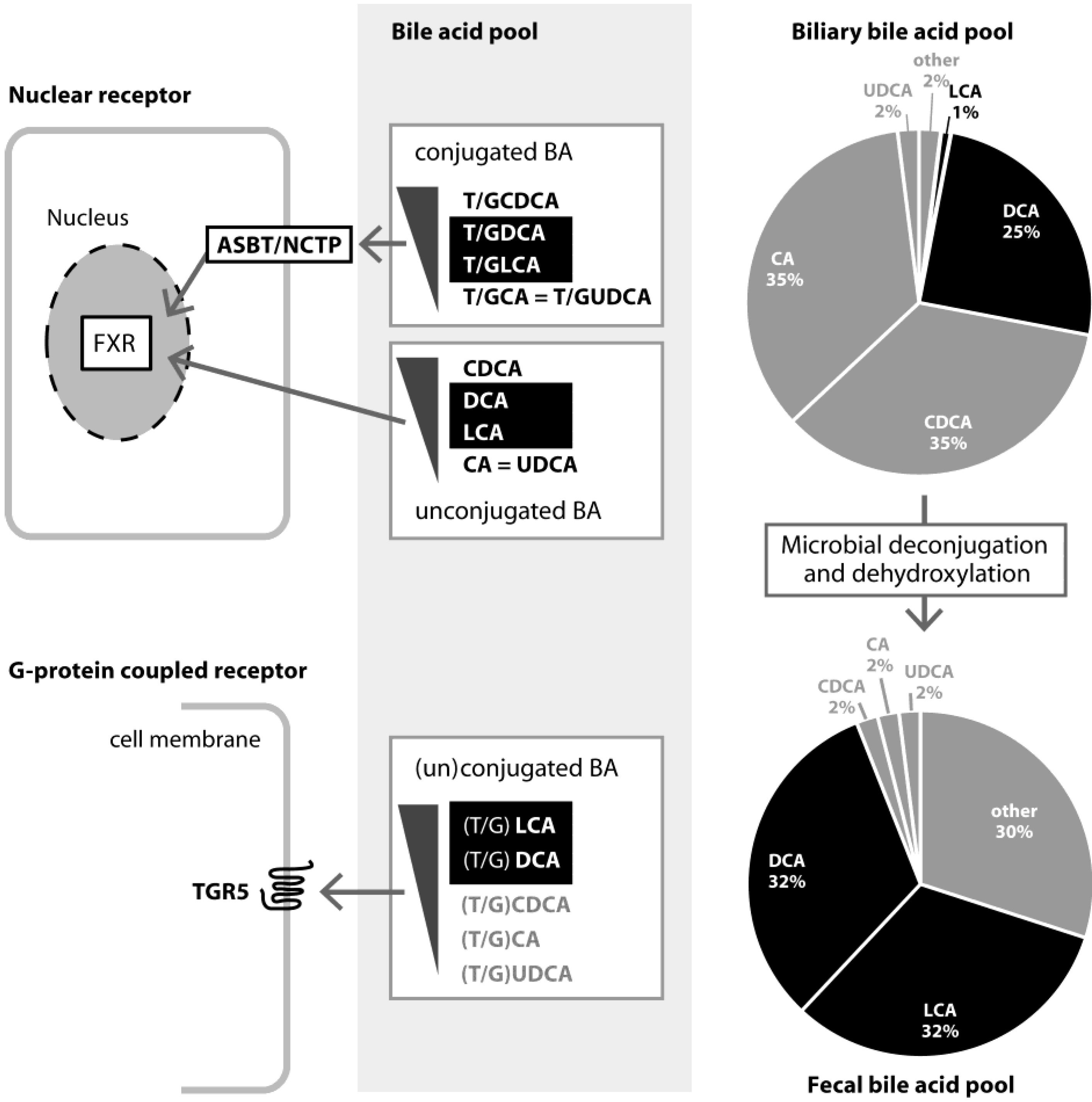
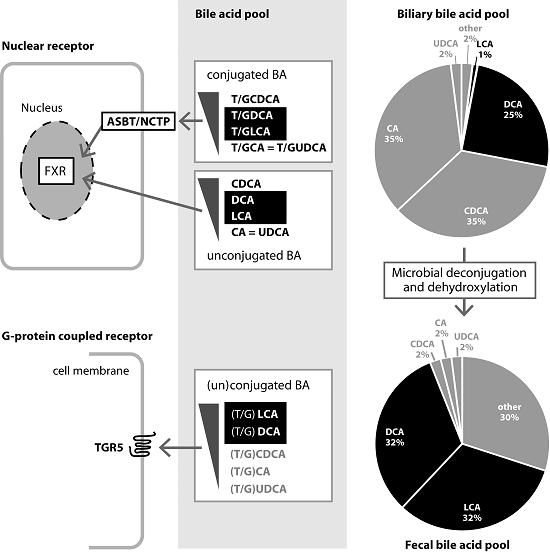
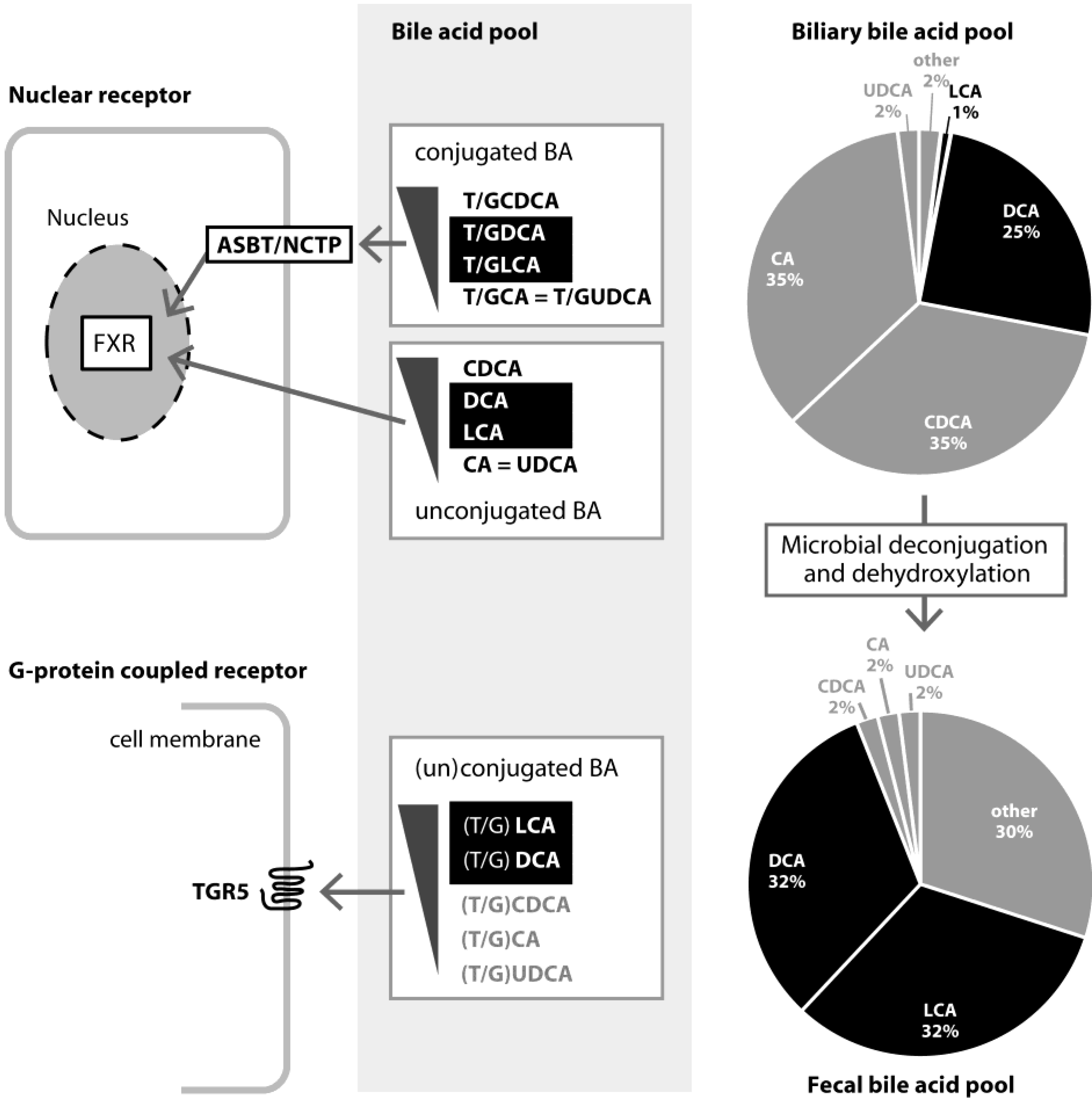
3. Role and Functions of FXR
3.1. FXR and Bile Acid Metabolism
3.2. Role of FXR in Metabolic Homeostasis
3.3. Role of FXR in Mucosal Protection
3.4. Role of FXR in Inflammation
4. Role and Functions of TGR5
4.1. TGR5 and BA Metabolism
4.2. Role of TGR5 in Metabolic Homeostasis
4.3. Role of TGR5 in Mucosal Protection
4.4. Role of TGR5 in inflammation
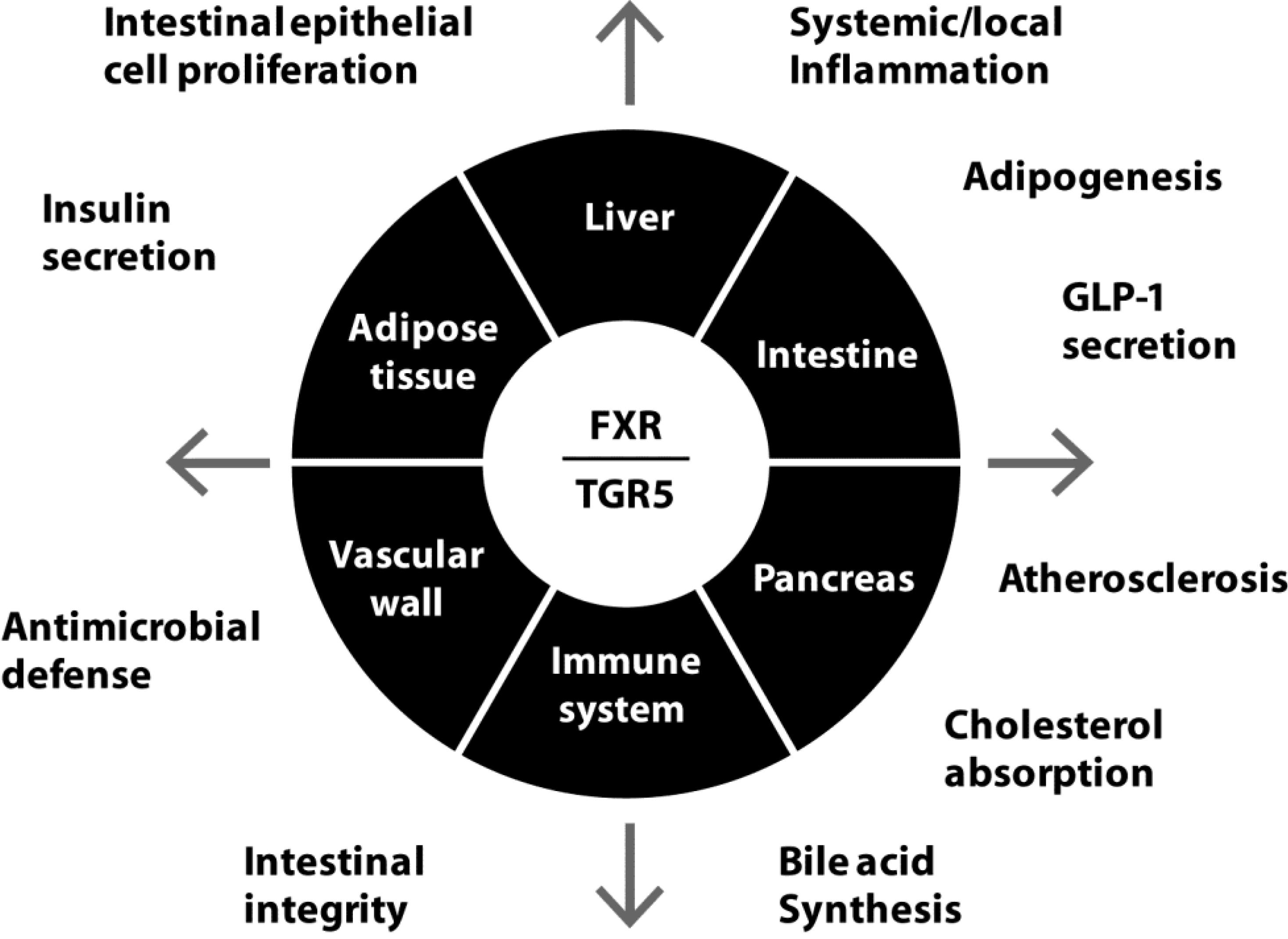
5. FXR and TGR5 as Therapeutic Targets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
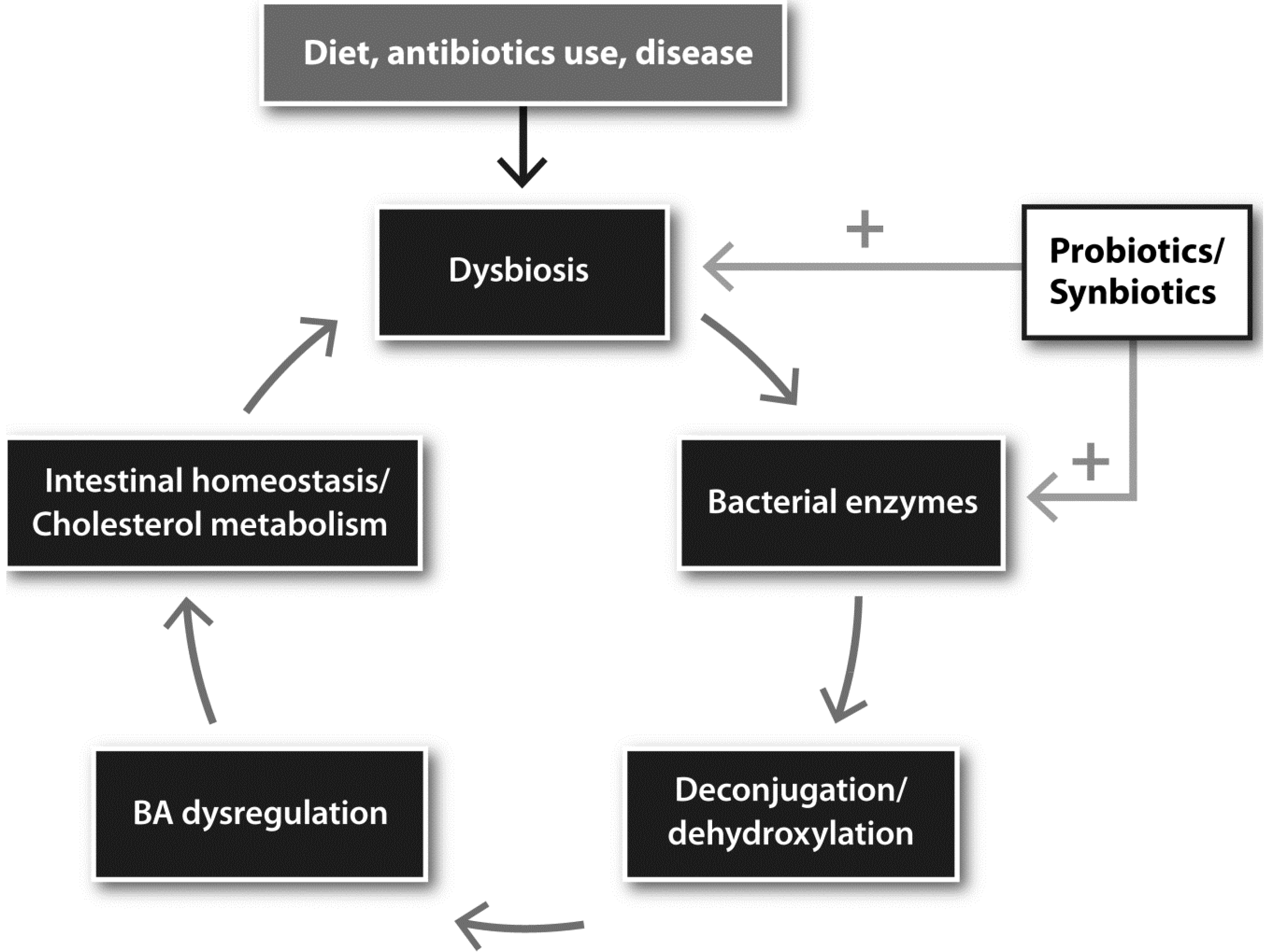
6. Dysbiosis, BA Dysregulation and Inflammatory Bowel Disease
7. Dysbiosis, BA Dysregulation and Metabolic Disease
8. Dietary Interventions with Pro or Synbiotics
9. Conclusions and Perspective

Author Contributions
Conflicts of Interest
References
- Scholtens, P.A.; Oozeer, R.; Martin, R.; Amor, K.B.; Knol, J. The early settlers: Intestinal microbiology in early life. Annu. Rev. Food Sci. Technol. 2012, 3, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Nat. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Rajilic-Stojanovic, M.; de Vos, W.M. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 2008, 57, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Knights, D.; Gonzalez, A.; Waldron, L.; Segata, N.; Knight, R.; Huttenhower, C.; Ley, R.E. A guide to enterotypes across the human body: Meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Smidt, H.; de Vos, W.M.; Belzer, C. The function of our microbiota: Who is out there and what do they do? Front. Cell. Infect. Microbiol. 2012, 2, 104. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Blumberg, R.S. Life at the beginning: Perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nature Immunol. 2014, 15, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Mshvildadze, M.; Neu, J. The infant intestinal microbiome: Friend or foe? Early Hum. Dev. 2010, 86, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Walujkar, S.A.; Dhotre, D.P.; Marathe, N.P.; Lawate, P.S.; Bharadwaj, R.S.; Shouche, Y.S. Characterization of bacterial community shift in human ulcerative colitis patients revealed by illumina based 16s rrna gene amplicon sequencing. Gut Pathog. 2014, 6, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Genetics and environmental interactions shape the intestinal microbiome to promote inflammatory bowel disease versus mucosal homeostasis. Gastroenterology 2010, 139, 1816–1819. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Licona-Limon, P.; Flavell, R.A. Role of the intestinal microbiome in liver disease. J. Autoimmun. 2013, 46, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Gerhold, K.; Stobberingh, E.E.; Thijs, C.; Zimmermann, K.; Lau, S.; Hamelmann, E. Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. J. Allergy Clin. Immunol. 2013, 132, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyoty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in european women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Artis, D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu. Rev. Immunol. 2010, 28, 623–667. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nature Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Rudensky, A.Y. The effects of commensal microbiota on immune cell subsets and inflammatory responses. Immunol. Rev. 2012, 245, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Patti, M.E. Bile acids, obesity, and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Charach, G.; Grosskopf, I.; Rabinovich, A.; Shochat, M.; Weintraub, M.; Rabinovich, P. The association of bile acid excretion and atherosclerotic coronary artery disease. Ther. Adv. Gastroenterol. 2011, 4, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Abrahamse, E.; Minekus, M.; van Aken, G.A.; van de Heijning, B.; Knol, J.; Bartke, N.; Oozeer, R.; van der Beek, E.M.; Ludwig, T. Development of the digestive system—Experimental challenges and approaches of infant lipid digestion. Food Dig. 2012, 3, 63–77. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nature Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nature Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. The enterohepatic circulation of bile acids in mammals: Form and functions. Front. Biosci. 2009, 14, 2584–2598. [Google Scholar] [CrossRef]
- Bortolini, O.; Medici, A.; Poli, S. Biotransformations on steroid nucleus of bile acids. Steroids 1997, 62, 564–577. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Joyce, S.A.; MacSharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Nat. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Pei, L.; Evans, R.M. Nuclear receptors: Decoding metabolic disease. FEBS Sett. 2008, 582, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell. Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Haywood, J.; Craddock, A.L.; Belinsky, M.G.; Kruh, G.D.; Dawson, P.A. The organic solute transporter alpha-beta, ostalpha-ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc. Nat. Acad. Sci. USA 2008, 105, 3891–3896. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, F.; Guo, G.L. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol. Res. 2011, 63, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid x receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Rainaldi, S.; Bovenga, F.; Murzilli, S.; Moschetta, A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep. 2014, 7, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PloS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Cariello, M.; Morgano, A.; Gross, I.; Vegliante, M.C.; Murzilli, S.; Salvatore, L.; Freund, J.N.; Sabba, C.; Moschetta, A. Transcriptional regulation of the intestinal nuclear bile acid farnesoid x receptor (FXR) by the caudal-related homeobox 2 (CDX2). J. Biol. Chem. 2014, 289, 28421–28432. [Google Scholar] [CrossRef] [PubMed]
- Kazumori, H.; Ishihara, S.; Rumi, M.A.; Kadowaki, Y.; Kinoshita, Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in barrett’s epithelium. Gut 2006, 55, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-D.; Chen, W.-D.; Moore, D.D.; Huang, W. FXR: A metabolic regulator and cell protector. Cell Res. 2008, 18, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Nat. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Huang, Y.; Yan, L.; Gao, M.; Liu, D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res. 2013, 30, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; van Harmelen, K.; Duran-Sandoval, D.; van Dijk, T.H.; Grefhorst, A.; Abdelkarim, M.; Caron, S.; Torpier, G.; Fruchart, J.-C.; Gonzalez, F.J. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J. Biol. Chem. 2006, 281, 11039–11049. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Horai, Y.; Houten, S.M.; Morimoto, K.; Sugizaki, T.; Arita, E.; Mataki, C.; Sato, H.; Tanigawara, Y.; Schoonjans, K. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J. Biol. Chem. 2011, 286, 26913–26920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, X.; Heemstra, L.A.; Chen, W.-D.; Xu, J.; Smith, J.L.; Ma, H.; Kasim, N.; Edwards, P.A.; Novak, C.M. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol. Endocrinol. 2012, 26, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Prawitt, J.; Abdelkarim, M.; Stroeve, J.H.; Popescu, I.; Duez, H.; Velagapudi, V.R.; Dumont, J.; Bouchaert, E.; Van Dijk, T.H.; Lucas, A. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes 2011, 60, 1861–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Stroeve, J.H.; Brufau, G.; Stellaard, F.; Gonzalez, F.J.; Staels, B.; Kuipers, F. Intestinal FXR-mediated FGF15 production contributes to diurnal control of hepatic bile acid synthesis in mice. Lab. Investig. 2010, 90, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.D. Bacterial translocation from the gastrointestinal tract. Trends Microbiol. 1995, 3, 149–154. [Google Scholar] [CrossRef]
- Slocum, M.M.; Sittig, K.M.; Specian, R.D.; Deitch, E.A. Absence of intestinal bile promotes bacterial translocation. Am. Surg. 1992, 58, 305–310. [Google Scholar] [PubMed]
- Ding, J.W.; Andersson, R.; Soltesz, V.; Willen, R.; Bengmark, S. The role of bile and bile acids in bacterial translocation in obstructive jaundice in rats. Eur. Surg. Res. 1993, 25, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Zuniga, V.; Bartoli, R.; Planas, R.; Hofmann, A.F.; Vinado, B.; Hagey, L.R.; Hernandez, J.M.; Mane, J.; Alvarez, M.A.; Ausina, V.; et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 2003, 37, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.M.; Knoefler, D.; Vitvitsky, V.; Banerjee, R.; Jakob, U. Bile salts act as effective protein-unfolding agents and instigators of disulfide stress in vivo. Proc. Nat. Acad. Sci. USA 2014, 111, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Nat. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Eckmann, L. How bile acids confer gut mucosal protection against bacteria. Proc. Nat. Acad. Sci. USA 2006, 103, 4333–4334. [Google Scholar] [CrossRef] [PubMed]
- Maran, R.R.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J. Pharmacol. Exp. Ther. 2009, 328, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.M.; Zhan, L.; Maru, D.; Shureiqi, I.; Pickering, C.R.; Kiriakova, G.; Izzo, J.; He, N.; Wei, C.; Baladandayuthapani, V.; et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am. J. Physiol. 2014, 306, G48–G58. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Ogawa, S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nature rev. Immunol. 2006, 6, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A sumoylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyan, N.; Luo, Y.; Sun, A.Q.; Suchy, F.J. Sumoylation of the farnesoid X receptor (FXR) regulates the expression of FXR target genes. J. Biol. Chem. 2013, 288, 13850–13862. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.; Siersema, P.D.; van Erpecum, K.J.; van Mil, S.W. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kB signaling in the intestine. Biochim. Biophys. Acta 2011, 1812, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, G.; Hommes, D.W.; de Jong, D.J.; Stokkers, P.C.; Verspaget, H.W.; Weersma, R.K.; van der Woude, C.J.; Stapelbroek, J.M.; Schipper, M.E.I.; Wijmenga, C.; et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PloS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.P.; Chen, Y.; Zimniak, P.; Cheng, K. Deoxycholic acid conjugates are muscarinic cholinergic receptor antagonists. Pharmacology 2002, 65, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, D.; Shen, W.; Dong, H.F.; Wang, J.M.; Oppenheim, J.J.; Howard, M.Z. Characterization of chenodeoxycholic acid as an endogenous antagonist of the G-coupled formyl peptide receptors. Inflamm. Res. 2000, 49, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (GPBAR1/M-BAR) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Holmstrom, S.R.; Kir, S.; Umetani, M.; Schmidt, D.R.; Kliewer, S.A.; Mangelsdorf, D.J. The G protein-coupled bile acid receptor, tgr5, stimulates gallbladder filling. Mol. Endocrinol. 2011, 25, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Tache, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liv. Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.P.; Rajagopal, S.; Mahavadi, S.; Mirshahi, F.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic β cells. Biochem. Biophys. Res. Commun. 2012, 427, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Kida, T.; Tsubosaka, Y.; Hori, M.; Ozaki, H.; Murata, T. Bile acid receptor TGR5 agonism induces no production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PloS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Kurita, C.; Nagao, T.; Usami, E.; Nakao, T.; Watanabe, S.; Kobayashi, J.; Yamazaki, F.; Tanaka, H.; Inagaki, N.; et al. Inhibition of tumor necrosis factor-α and interleukin-1-β production by β-adrenoceptor agonists from lipopolysaccharide-stimulated human peripheral blood mononuclear cells. Pharmacology 1997, 54, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki-Anzai, S.; Masuda, M.; Levi, M.; Keenan, A.L.; Miyazaki, M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PloS ONE 2014, 9. [Google Scholar] [CrossRef]
- Wall, E.A.; Zavzavadjian, J.R.; Chang, M.S.; Randhawa, B.; Zhu, X.; Hsueh, R.C.; Liu, J.; Driver, A.; Bao, X.R.; Sternweis, P.C.; et al. Suppression of LPS-induced TNF-α production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Ma, X.; Fu, X.; Meng, Z.; Zhang, W.; Wang, Y.D.; Van Ness, C.; Yu, D.; Xu, R.; Huang, W. GPBAR1/TGR5 mediates bile acid-induced cytokine expression in murine kupffer cells. PloS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Pean, N.; Doignon, I.; Garcin, I.; Besnard, A.; Julien, B.; Liu, B.; Branchereau, S.; Spraul, A.; Guettier, C.; Humbert, L.; et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology 2013, 58, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated b cells (Nf-kB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Pols, T.W.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J. Clin.Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acid signaling in liver metabolism and diseases. J. Lipid 2011, 2012. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nature Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Dore, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, C.; Tang, C.; Li, N.; Li, J. Molecular-phylogenetic characterization of the microbiota in ulcerated and non-ulcerated regions in the patients with crohn’s disease. PloS ONE 2012, 7. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Dore, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Nat. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [PubMed]
- Labbe, A.; Ganopolsky, J.G.; Martoni, C.J.; Prakash, S.; Jones, M.L. Bacterial bile metabolising gene abundance in crohn’s, ulcerative colitis and type 2 diabetes metagenomes. PloS ONE 2014, 9. [Google Scholar] [CrossRef]
- Attinkara, R.; Mwinyi, J.; Truninger, K.; Regula, J.; Gaj, P.; Rogler, G.; Kullak-Ublick, G.A.; Eloranta, J.J.; Swiss, I.B.D.C.S.G. Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor fxr, with inflammatory bowel disease. BMC Res. Note 2012, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, K.D.; Hanson, M.A. The developmental origins, mechanisms, and implications of metabolic syndrome. J. Nutr 2010, 140, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M. Maternal regulation of fetal development and health in adult life. Eur. J. Obstet. Gynecol. Reprod. Biol. 1998, 78, 141–150. [Google Scholar] [CrossRef]
- Barker, D.J.; Gluckman, P.D.; Godfrey, K.M.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef]
- Cleal, J.K.; Poore, K.R.; Boullin, J.P.; Khan, O.; Chau, R.; Hambidge, O.; Torrens, C.; Newman, J.P.; Poston, L.; Noakes, D.E.; et al. Mismatched pre- and postnatal nutrition leads to cardiovascular dysfunction and altered renal function in adulthood. Proc. Nat. Acad. Sci. USA 2007, 104, 9529–9533. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Breier, B.H.; Cutfield, W.S.; Hofman, P.L.; Gluckman, P.D. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol.. Endocrinol. Metab. 2000, 279, 83–87. [Google Scholar]
- DiBaise, J.K.; Zhang, H.; Crowell, M.D.; Krajmalnik-Brown, R.; Decker, G.A.; Rittmann, B.E. Gut microbiota and its possible relationship with obesity. Mayo Clin. Proc. 2008, 83, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Xu, Z.; Knight, R. Meta-analyses of human gut microbes associated with obesity and ibd. FEBS Lett. 2014, 588, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, L.; van Hoorenbeeck, K.; Kortleven, I.; van Noten, C.; Hens, N.; Vael, C.; Goossens, H.; Desager, K.N.; Vankerckhoven, V. Differences in gut microbiota composition between obese and lean children: A cross-sectional study. Gut Pathog. 2013, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tims, S.; Derom, C.; Jonkers, D.M.; Vlietinck, R.; Saris, W.H.; Kleerebezem, M.; de Vos, W.M.; Zoetendal, E.G. Microbiota conservation and bmi signatures in adult monozygotic twins. ISME J. 2013, 7, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and scfa in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottiere, H.M.; Raes, J.; Ehrlich, D.; Dore, J. A metagenomic insight into our gut's microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Damms-Machado, A.; Mitra, S.; Schollenberger, A.E.; Kramer, K.M.; Meile, T.; Königsrainer, A.; Huson, D.H.; Bischoff, S.C. Effects of surgical and dietary weight loss therapy for obesity on gut microbiota composition and nutrient absorption. Bio. Med. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Flynn, C.R.; Albaugh, V.L.; Cai, S.; Cheung-Flynn, J.; Williams, P.E.; Brucker, R.M.; Bordenstein, S.R.; Guo, Y.; Wasserman, D.H.; Abumrad, N.N. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nature Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Tremaroli, V.; Clemmensen, C.; Kovatcheva-Datchary, P.; Myronovych, A.; Karns, R.; Wilson-Pérez, H.E.; Sandoval, D.A.; Kohli, R.; Bäckhed, F. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014, 509, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Out, C.; Fuentes, S.; Jonker, L.; Reuling, I.; Kootte, R.S.; van Nood, E.; Holleman, F.; Knaapen, M.; Romijn, J.A. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J. Hepatol. 2014, 60, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nature commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.Y.; Sun, W.W.; Ma, Y.; Zhu, H.; Yang, P.; Wei, H.; Zeng, B.H.; Zhang, Q.; Liu, Y.; Li, W.X.; et al. Microbiota prevents cholesterol loss from the body by regulating host gene expression in mice. Sci. Rep. 2015, 5, 10512. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.D. The quantification of cholesterol excretion and degradation in the isotopic steady state in the rat: The influence of dietary cholesterol. J. Lipid Res. 1964, 5, 409–417. [Google Scholar] [PubMed]
- Wostmann, B.S.; Wiech, N.L.; Kung, E. Catabolism and elimination of cholesterol in germfree rats. J. Lipid Res 1966, 7, 77–82. [Google Scholar] [PubMed]
- Jones, M.L.; Tomaro-Duchesneau, C.; Martoni, C.J.; Prakash, S. Cholesterol lowering with bile salt hydrolase-active probiotic bacteria, mechanism of action, clinical evidence, and future direction for heart health applications. Expert Opin. Biol. Ther. 2013, 13, 631–642. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V. Use of probiotics to correct dysbiosis of normal microbiota following disease or disruptive events: A systematic review. BMJ Open 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Doesburg, K.; Iwasaki, T.; Mierau, I. Screening of lactic acid bacteria for bile salt hydrolase activity. Journal of dairy science 1999, 82, 2530–2535. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan, C.G. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Rainteau, D.; Rajca, S.; Humbert, L.; Farabos, D.; Maubert, M.; Grondin, V.; Jouet, P.; Bouhassira, D.; Seksik, P.; et al. Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol. Motil. 2012, 24, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Richards, D.M.; Rahimi, E.F.; Krill, J.T.; Jelinek, K.A.; DuPont, A.W. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin. Exp. Gastroenterol. 2014, 7, 473–487. [Google Scholar] [PubMed]
- Saez-Lara, M.J.; Gomez-Llorente, C.; Plaza-Diaz, J.; Gil, A. The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: A systematic review of randomized human clinical trials. Bio. Med. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Stojancevic, M.; Stankov, K.; Mikov, M. The impact of farnesoid x receptor activation on intestinal permeability in inflammatory bowel disease. Can. J. Gastroenterol. 2012, 26, 631–637. [Google Scholar] [PubMed]
- Van Schaik, F.D.; Gadaleta, R.M.; Schaap, F.G.; van Mil, S.W.; Siersema, P.D.; Oldenburg, B.; van Erpecum, K.J. Pharmacological activation of the bile acid nuclear farnesoid X receptor is feasible in patients with quiescent crohn’s colitis. PloS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Letter to the editor regarding the report of duboc et al: Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel disease. Gut 2013, 62, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Cholesterol lowering and inhibition of sterol absorption by lactobacillus reuteri ncimb 30242: A randomized controlled trial. Eur. J. Clin. Nutr. 2012, 66, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.; Stankov, K.; Mikov, M. Probiotics—Interactions with bile acids and impact on cholesterol metabolism. Appl. Biochem. Biotechnol. 2012, 168, 1880–1895. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. The function of bile salts in fat absorption. The solvent properties of dilute micellar solutions of conjugated bile salts. Biochem. J. 1963, 89, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Angelakis, E.; Merhej, V.; Raoult, D. Related actions of probiotics and antibiotics on gut microbiota and weight modification. Lancet. Infect. Dis. 2013, 13, 889–899. [Google Scholar] [CrossRef]
- Sipka, S.; Bruckner, G. The immunomodulatory role of bile acids. Int. Arch. Allergy Immunol. 2014, 165, 1–8. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baars, A.; Oosting, A.; Knol, J.; Garssen, J.; Van Bergenhenegouwen, J. The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5. Microorganisms 2015, 3, 641-666. https://doi.org/10.3390/microorganisms3040641
Baars A, Oosting A, Knol J, Garssen J, Van Bergenhenegouwen J. The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5. Microorganisms. 2015; 3(4):641-666. https://doi.org/10.3390/microorganisms3040641
Chicago/Turabian StyleBaars, Annemarie, Annemarie Oosting, Jan Knol, Johan Garssen, and Jeroen Van Bergenhenegouwen. 2015. "The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5" Microorganisms 3, no. 4: 641-666. https://doi.org/10.3390/microorganisms3040641
APA StyleBaars, A., Oosting, A., Knol, J., Garssen, J., & Van Bergenhenegouwen, J. (2015). The Gut Microbiota as a Therapeutic Target in IBD and Metabolic Disease: A Role for the Bile Acid Receptors FXR and TGR5. Microorganisms, 3(4), 641-666. https://doi.org/10.3390/microorganisms3040641