Conservation Implications of Shifting Gut Microbiomes in Captive-Reared Endangered Voles Intended for Reintroduction into the Wild

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Sample Collection
2.3. Sample Analysis
2.4. Statistical Analysis
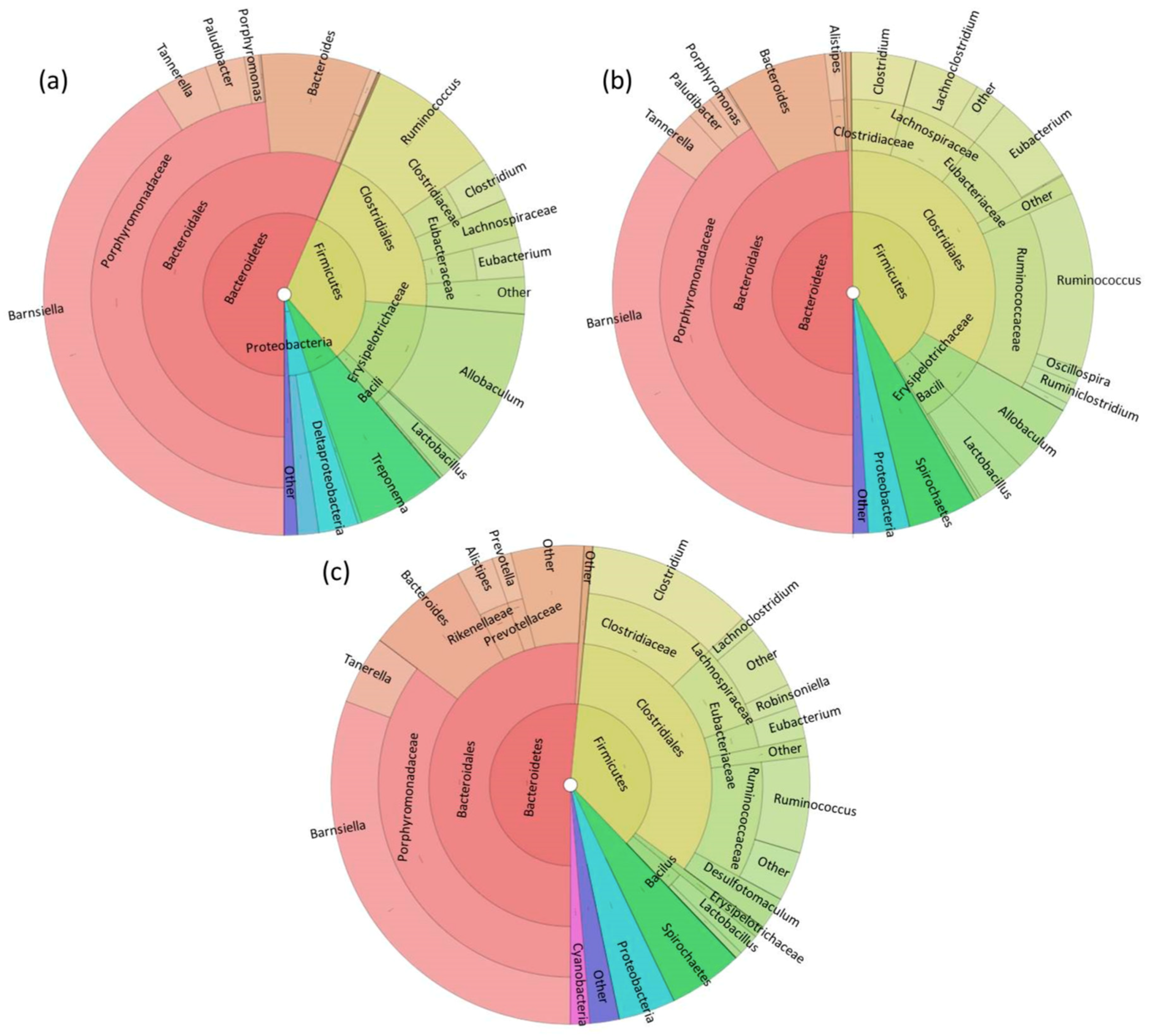
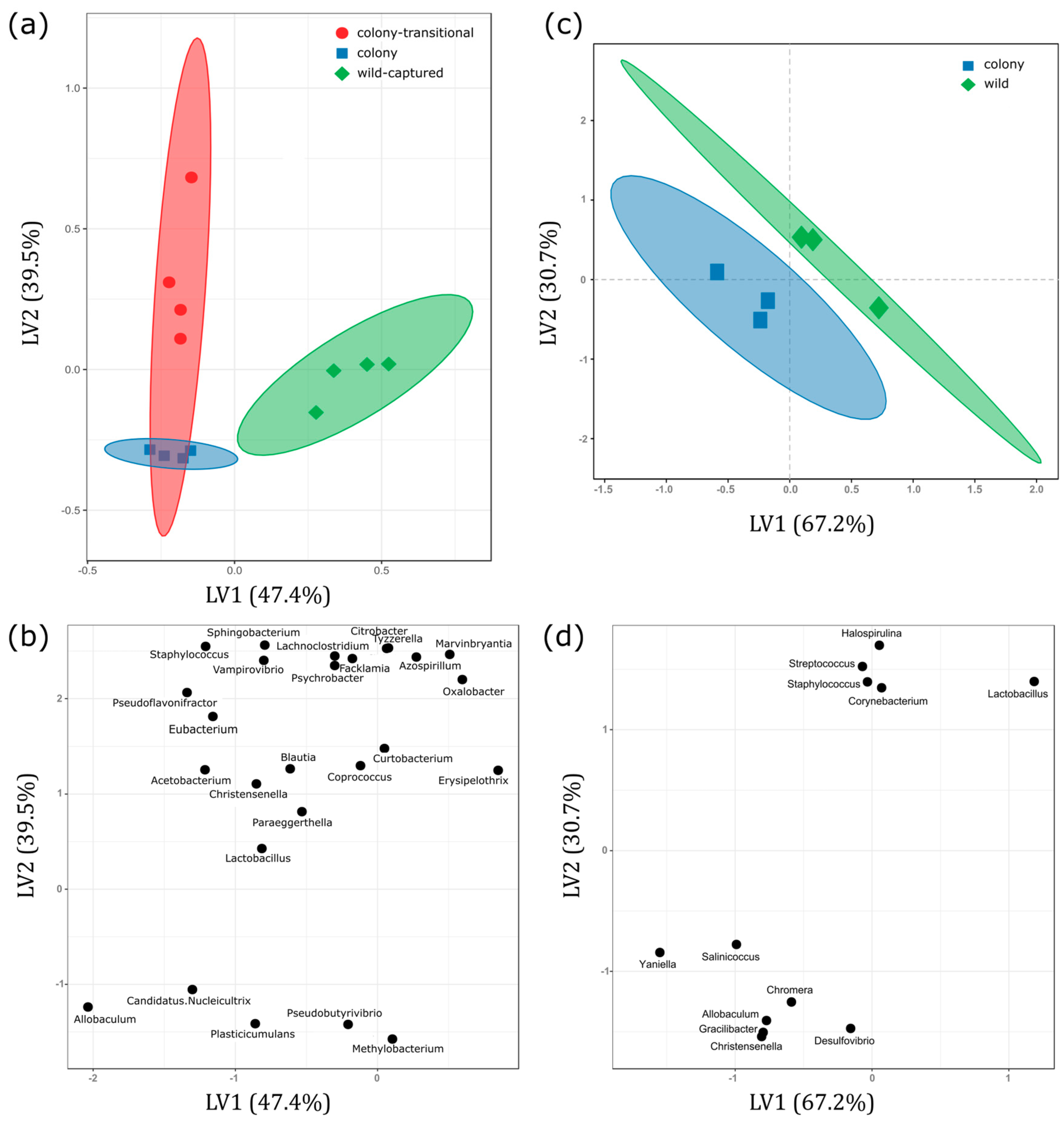
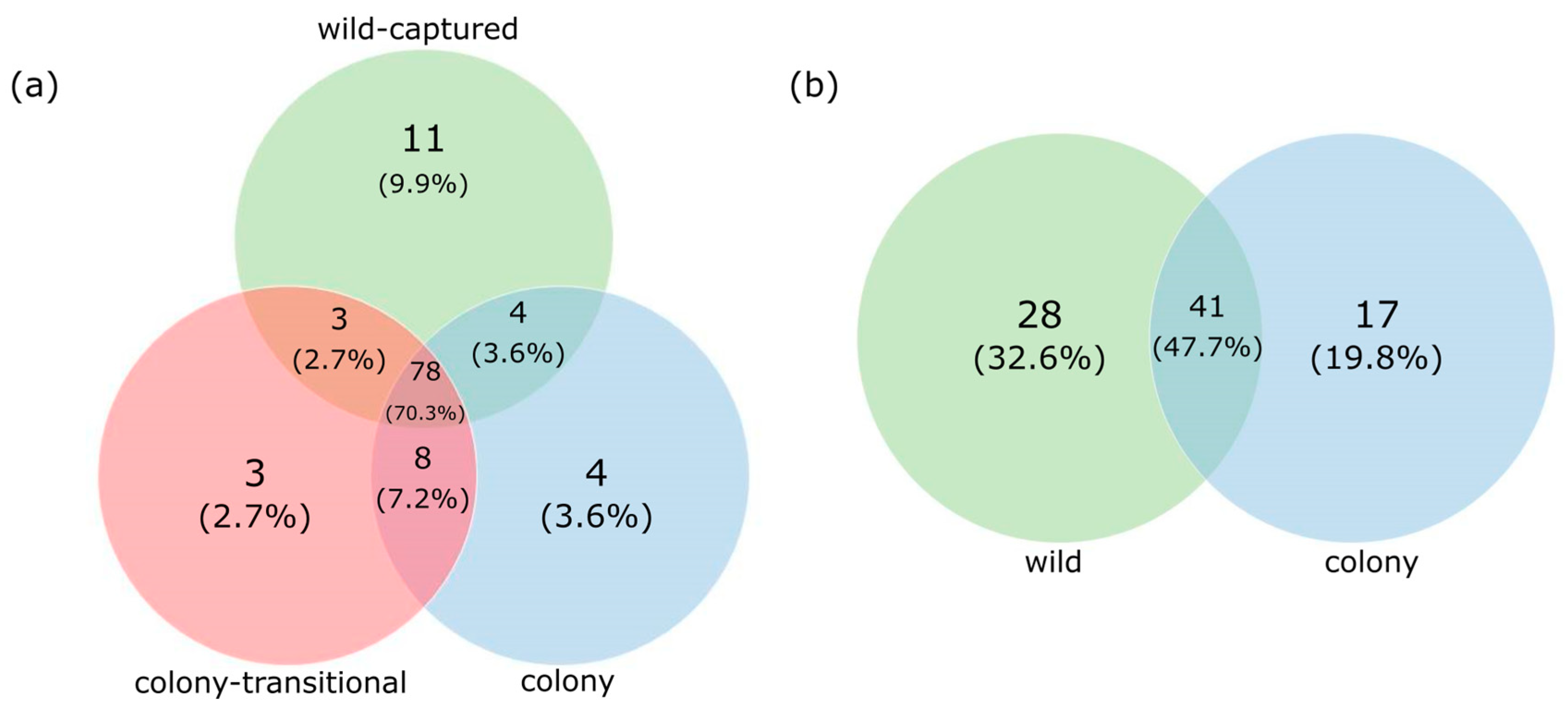
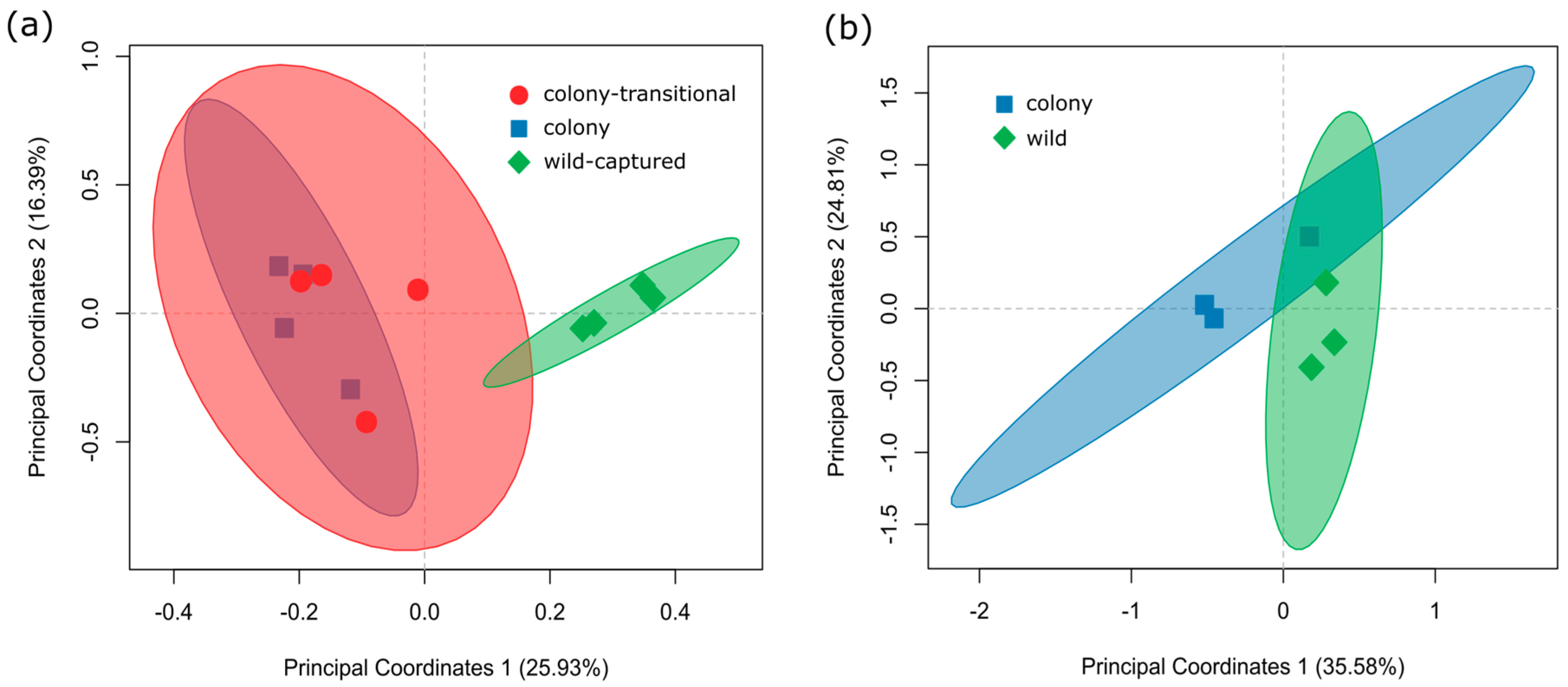
3. Results
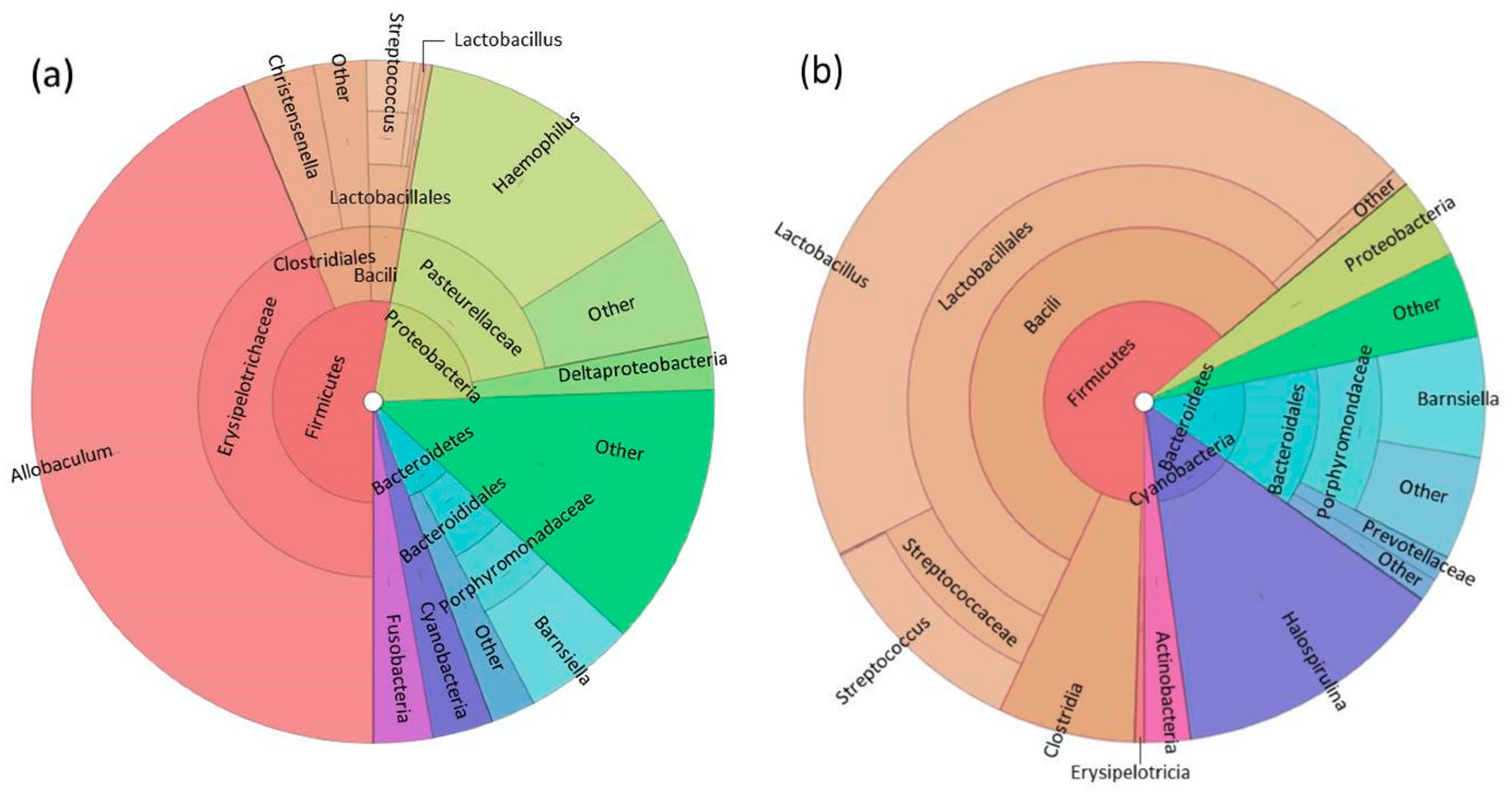
3.1. Fecal Microbiome Analysis
3.2. Foregut Microbiome Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Cudworth, N.L.; Koprowski, J.L. Microtus californicus (Rodentia: Cricetidae). Mamm. Species 2010, 42, 230–243. [Google Scholar] [CrossRef]
- U.S. Fish and Wildlife Service. Amargosa Voles (Microtus californicus scirpensis) Recovery Plan; U.S. Fish and Wildlife Service: Portland, OR, USA, 1997.
- California Department of Fish and Game. Distribution and Abundance of the Amargosa vole (Microtus californicus scirpensis); California Department of Fish and Game: Sacramento, CA, USA, 1998.
- Neuwald, J.L. Population isolation exacerbates conservation genetic concerns in the endangered Amargosa vole, Microtus californicus scirpensis. Biol. Conserv. 2010, 143, 2028–2038. [Google Scholar] [CrossRef]
- California Department of Fish and Game. At the Crossroads: A Report on the Status of California’s Endangered and Rare Fish and Wildlife; California Department of Fish and Game: Sacramento, CA, USA, 1980.
- Nowak, R. Endangered and threatened wildlife and plants; determination of endangered status and critical habitat for the Amargosa vole. Final Rule Fed. Reg. 1984, 49, 45160–45163. [Google Scholar]
- California Department of Fish and Wildlife. Amargosa vole recovery permit TE54614A-1 activity report: October 2014–December 2015; California Department of Fish and Game: Sacramento, CA, USA, 2016.
- Allan, N.; Pesapane, R.; Foley, J.; Clifford, D. Successful care and propagation of the endangered Amargosa vole (Microtus californicus scirpensis) in captivity. Zoo Biol. 2018, 37, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Bright, P.; Morris, P. Animal translocation for conservation: Performance of dormice in relation to release methods, origin and season. J. Appl. Ecol. 1994, 31, 699–708. [Google Scholar] [CrossRef]
- Mathews, F.; Orros, M.; McLaren, G.; Gelling, M.; Foster, R. Keeping fit on the ark: Assessing the suitability of captive-bred animals for release. Biol. Conserv. 2005, 121, 569–577. [Google Scholar] [CrossRef]
- Biggins, D.E.; Godbey, J.L.; Matchett, M.R.; Livieri, T.M.; Roelle, J.; Miller, B. Habitat preferences and intraspecific competition in black-footed ferrets. Recovery of the Black-footed Ferret: Progress and Continuing Challenges. In Proceedings of the Symposium on the Status of the Black-Footed Ferret and Its Habitat, Fort Collins, CO, USA, 28–29 September 2004; pp. 129–140. [Google Scholar]
- Brown, J.L.; Collopy, M.W.; Gott, E.J.; Juergens, P.W.; Montoya, A.B.; Hunt, W.G. Wild-reared Aplomado falcons survive and recruit at higher rates than hacked falcons in a common environment. Biol. Conserv. 2006, 131, 453–458. [Google Scholar] [CrossRef]
- Jule, K.R.; Leaver, L.A.; Lea, S.E. The effects of captive experience on reintroduction survival in carnivores: A review and analysis. Biol. Conserv. 2008, 141, 355–363. [Google Scholar] [CrossRef]
- O’Regan, H.J.; Kitchener, A.C. The effects of captivity on the morphology of captive, domesticated and feral mammals. Mamm. Rev. 2005, 35, 215–230. [Google Scholar] [CrossRef]
- Snyder, N.F.; Derrickson, S.R.; Beissinger, S.R.; Wiley, J.W.; Smith, T.B.; Toone, W.D.; Miller, B. Limitations of captive breeding in endangered species recovery. Conserv. Biol. 1996, 10, 338–348. [Google Scholar] [CrossRef]
- Scott, M.E. The impact of infection and disease on animal populations: Implications for conservation biology. Conserv. Biol. 1988, 2, 40–56. [Google Scholar] [CrossRef]
- Viggers, K.; Lindenmayer, D.; Spratt, D. The importance of disease in reintroduction programmes. Wildl. Res. 1993, 20, 687–698. [Google Scholar] [CrossRef]
- Kleiman, D.G. Reintroduction of captive mammals for conservation. BioScience 1989, 39, 152–161. [Google Scholar] [CrossRef]
- California Department of Fish and Wildlife. Annual Performance Report: Captive Breeidng of a Model Species and Reconstruction Habitat Ahead of Translocation of the Critically Endangered Amargosa vole—F14AP01006. May 2015–June 2016; California Department of Fish and Wildlife: Sacramento, CA, USA, 2016.
- Hammond, K.A.; Wunder, B.A. The role of diet quality and energy need in the nutritional ecology of a small herbivore, Microtus ochrogaster. Physiol. Zool. 1991, 64, 541–567. [Google Scholar] [CrossRef]
- Lovegrove, B.G. The allometry of rodent intestines. J. Comp. Physiol. B 2010, 180, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M.; Frey, R.; Kiefer, B.; Lechner-Doll, M.; Loehlein, W.; Polster, C.; Rössner, G.; Streich, W.J. The maximum attainable body size of herbivorous mammals: Morphophysiological constraints on foregut, and adaptations of hindgut fermenters. Oecologia 2003, 136, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Hume, I.D.; Morgan, K.R.; Kenagy, G. Digesta retention and digestive performance in sciurid and microtine rodents: Effects of hindgut morphology and body size. Physiol. Zool. 1993, 66, 396–411. [Google Scholar] [CrossRef]
- Justice, K.E.; Smith, F.A. A model of dietary fiber utilization by small mammalian herbivores, with empirical results for Neotoma. Am. Nat. 1992, 139, 398–416. [Google Scholar] [CrossRef]
- McBee, R.H. Significance of intestinal microflora in herbivory. Annu. Rev. Ecol. Syst. 1971, 2, 165–176. [Google Scholar] [CrossRef]
- Morrison, M.; Pope, P.B.; Denman, S.E.; McSweeney, C.S. Plant biomass degradation by gut microbiomes: More of the same or something new? Curr. Opin. Biotechnol. 2009, 20, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, E.D.; Sonnenburg, J.L. Starving our microbial self: The deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014, 20, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.M.; Rogers, T.L.; Carlini, A.R.; Brown, M.V. Diet and phylogeny shape the gut microbiota of Antarctic seals: A comparison of wild and captive animals. Environ. Microbiol. 2013, 15, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Scupham, A.J.; Patton, T.G.; Bent, E.; Bayles, D.O. Comparison of the cecal microbiota of domestic and wild turkeys. Microb. Ecol. 2008, 56, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Xenoulis, P.G.; Gray, P.L.; Brightsmith, D.; Palculict, B.; Hoppes, S.; Steiner, J.M.; Tizard, I.; Suchodolski, J.S. Molecular characterization of the cloacal microbiota of wild and captive parrots. Vet. Microbiol. 2010, 146, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Dearing, M.D. Wild-caught rodents retain a majority of their natural gut microbiota upon entrance into captivity. Environ. Microbiol. Rep. 2014, 6, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Skopec, M.M.; Dearing, M.D. Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv. Physiol. 2014, 2, cou009. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Oliveira, R.A.; Djukovic, A.; Ubeda, C.; Xavier, K.B. Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep. 2015, 10, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Gomez, A.; Petrželková, K.J.; Whittier, C.A.; Todd, A.F.; Yeoman, C.J.; Nelson, K.E.; Wilson, B.A.; Stumpf, R.M.; Modrý, D. Effect of antibiotic treatment on the gastrointestinal microbiome of free-ranging western lowland gorillas (Gorilla g. gorilla). Microb. Ecol. 2016, 72, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Imai, D.M.; Pesapane, R.; Conroy, C.J.; Alarcón, C.N.; Allan, N.; Okino, R.A.; Fung, J.; Murphy, B.G.; Verstraete, F.J.; Foley, J.E. Apical elongation of molar teeth in captive Microtus voles. Vet. Pathol. 2018, 0300985818758469. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 10 August 2018).
- Kucheryavskiy, S. mdatools: Multivariate Data Analysis for Chemometrics; R Package Version 0.8.2; 2017.
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Amato, K.R.; Leigh, S.R.; Kent, A.; Mackie, R.I.; Yeoman, C.J.; Stumpf, R.M.; Wilson, B.A.; Nelson, K.E.; White, B.A.; Garber, P.A. The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microb. Ecol. 2015, 69, 434–443. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, Y.; Koren, O.; Spor, A.; LeDuc, C.; Gutman, R.; Stombaugh, J.; Knight, R.; Ley, R.E.; Leibel, R.L. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 2012, 20, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Ringø, E.; Sperstad, S.; Myklebust, R.; Refstie, S.; Krogdahl, Å. Characterisation of the microbiota associated with intestine of Atlantic cod (Gadus morhua L.): The effect of fish meal, standard soybean meal and a bioprocessed soybean meal. Aquaculture 2006, 261, 829–841. [Google Scholar] [CrossRef]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Van Tuan, B.; Van Minh, V. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 10376–10381. [Google Scholar] [CrossRef] [PubMed]
- Holscher, H.D.; Caporaso, J.G.; Hooda, S.; Brulc, J.M.; Fahey, G.C., Jr.; Swanson, K.S. Fiber supplementation influences phylogenetic structure and functional capacity of the human intestinal microbiome: Follow-up of a randomized controlled trial. Am. J. Clin. Nutr. 2014, 101, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Simpson, H.L.; Campbell, B.J. Review article: Dietary fibre-microbiota interactions. Aliment. Pharm. Ther. 2015, 42, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Weldon, L.; Abolins, S.; Lenzi, L.; Bourne, C.; Riley, E.M.; Viney, M. The gut microbiota of wild mice. PLoS ONE 2015, 10, e0134643. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Lambiase, A. The Family Sphingobacteriaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Germany, 2014. [Google Scholar]
- Magli, A.; Messmer, M.; Leisinger, T. Metabolism of Dichloromethane by the strict anaerobe Dehalobacterium formicoaceticum. Appl. Environ. Microbiol. 1998, 64, 646–650. [Google Scholar] [PubMed]
- Moore, L.V.H.; Moore, W.E.C. Oribaculum catoniae gen. nov., sp. nov.; Catonella morbi gen. nov., sp. nov.; Hallella sergens gen. nov., sp. nov.; Johnsonella ignava gen. nov., sp. nov.; and Dialister pneumosintes gen. nov., comb. nov., nom. Rev., anaerobic gram-negative Bacilli from the human gingival crevice. Int. J. Syst. Bacteriol. 1994, 44, 187–192. [Google Scholar] [PubMed]
- Schouw, A.; Eide, T.L.; Stokke, R.; Pedersen, R.B.; Steen, I.H.; Bodtker, G. Abyssivirga alkaniphila gen. nov., sp. nov., an alkane-degrading, anaerobic bacterium from a deep-sea hydrothermal vent system, and emended descriptions of Natranaerovirga pectinivora and Natranaerovirga hydrolytica. Int. J. Syst. Evol. Microbiol. 2016, 66, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Shivaji, S.; Ray, M.K.; Rao, N.S.; Saisree, L.; Jagannadham, M.V.; Kumar, G.S.; Reddy, G.S.N.; Bhargava, P.M. Sphingobacterium antarcticus sp. nov., a psychrotrophic bacterium from the soils of Schirmacher Oasis, Antarctica. Int. J. Syst. Bacteriol. 1992, 42, 102–106. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Tourova, T.P.; Panteleeva, A.N.; Kaparullina, E.N.; Muyzer, G. Anaerobic utilization of pectinous substrates at extremely haloalkaline conditions by Natranaerovirga pectinivora gen. nov., sp. nov., and Natranaerovirga hydrolytica sp. nov., isolated from hypersaline soda lakes. Extremophiles 2012, 16, 307–315. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Wüst, P.K.; Horn, M.A.; Drake, H.L. Clostridiaceae and Enterobacteriaceae as active fermenters in earthworm gut content. ISME J. 2011, 5, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wu, Q.; Dai, J.; Zhang, S.; Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 17714–17719. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Kohl, K.D.; Sadowska, E.T.; Rudolf, A.M.; Dearing, M.D.; Koteja, P. Experimental evolution on a wild mammal species results in modifications of gut microbial communities. Front. Microbiol. 2016, 7, 634. [Google Scholar] [CrossRef] [PubMed]
- Thoetkiattikul, H.; Mhuantong, W.; Laothanachareon, T.; Tangphatsornruang, S.; Pattarajinda, V.; Eurwilaichitr, L.; Champreda, V. Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing. Curr. Microbiol. 2013, 67, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Varel, V.; Yen, J.T. Microbial perspective on fiber utilization by swine. J. Anim. Sci. 1997, 75, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Borbón-García, A.; Reyes, A.; Vives-Flórez, M.; Caballero, S. Captivity shapes the gut microbiota of Andean bears: Insights into health surveillance. Front. Microbiol. 2017, 8, 1316. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Ruan, R.; McLaughlin, R.W.; Hao, Y.; Zheng, J.; Wang, D. Fecal bacterial composition of the endangered Yangtze finless porpoises living under captive and semi-natural conditions. Curr. Microbiol. 2016, 72, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Bache, R.; Pfennig, N. Selective isolation of Acetobacterium woodii on methoxylated aromatic acids and determination of growth yields. Arch. Microbiol. 1981, 130, 255–261. [Google Scholar] [CrossRef]
- Balch, W.E.; Schoberth, S.; Tanner, R.S.; Wolfe, R.S. Acetobacterium, a new genus of hydrogen-oxidizing, carbon dioxide-reducing, anaerobic bacteria. Int. J. Syst. Bacteriol. 1977, 27, 355–361. [Google Scholar] [CrossRef]
- Coder, D.M.; Goff, L.J. The host range of the chlorellavorous bacterium (“Vampirovibrio chlorellovorus”). J. Phycol. 1986, 46, 543–547. [Google Scholar] [CrossRef]
- Steenhoudt, O.; Vanderleyden, J. Azospirillum, a free-living nitrogen-fixing bacterium closely associated with grasses: Genetic, biochemical and ecological aspects. FEMS Microbiol. Rev. 2000, 24, 487–506. [Google Scholar] [CrossRef] [PubMed]
- Veraldi, S.; Gergenti, V.; Dassoni, F.; Gianotti, R. Erysipeloid: A review. Clin. Exp. Dermatol. 2009, 34, 859–886. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, H. The Order Cytophagales. In The Prokaryotes; Balows, A., Truper, H.G., Dworkin, M., Harder, W., Schleifer, K.M., Eds.; Springer: New York, NY, USA, 1992; pp. 3631–3675. [Google Scholar]
- Xie, G.; Bruce, D.C.; Challacombe, J.F.; Chertkov, O.; Detter, J.C.; Gilna, P.; Han, C.S.; Lucas, S.; Misra, M.; Myers, G.L.; et al. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 2007, 73, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Ren, N.; Li, Q.; Lin, M.; Wang, A.; Zhao, L. Ethanoligenens harbinese gen. nov., sp. nov., isolated from molasses water. Int. J. Syst. Evol. Microbiol. 2006, 56, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Leadbetter, J.R.; Breznak, J.A. Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Appl. Environ. Microbiol. 1996, 62, 3620–3631. [Google Scholar] [PubMed]
- Lin, C.; Miller, T.L. Phylogenetic analysis of Methanobrevibacter isloated from feces of humans and other animals. Clin. Exp. Dermatol. 1998, 169, 397–403. [Google Scholar]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M.J.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Hammond, K.A. The Role of Diet Quality and Energy Need in the Nutritional Ecology of a Small Herbivore. Ph.D. Thesis, Colorado State University, Fort Collins, CO, USA, 1989. [Google Scholar]
- Young Owl, M.; Batzli, G.O. The integrated processing response of voles to fibre content of natural diets. Funct. Ecol. 1998, 12, 4–13. [Google Scholar] [CrossRef]
- Greetham, H.L.; Gibson, G.R.; Giffard, C.; Hippe, H.; Merkhoffer, B.; Steiner, U.; Falsen, E.; Collins, M.D. Allobaculum stercoricanis gen. nov., sp. nov., isolated from canine feces. Anaerobe 2004, 10, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Dearing, M. Experience matters: Prior exposure to plant toxins enhances diversity of gut microbes in herbivores. Ecol. Lett. 2012, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Miller, A.W.; Marvin, J.E.; Mackie, R.; Dearing, M.D. Herbivorous rodents (Neotoma spp.) harbour abundant and active foregut microbiota. Environ. Microbiol. 2014, 16, 2869–2878. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, A.; Uchida, E.; Shichijo, H.; Sakamoto, S.H.; Morita, T.; Koshimoto, C. Microbial diversity in forestomach and caecum contents of the greater long-tailed hamster Tscherskia triton (Rodentia: Cricetidae). Mamm. Biol. 2016, 81, 46–52. [Google Scholar] [CrossRef]
- Barrangou, R.; Azcarate-Peril, M.A.; Duong, T.; Conners, S.B.; Kelly, R.M.; Klaenhammer, T.R. Global analysis of carbohydrate utilization by Lactobacillus acidophilus using cDNA microarrays. Proc. Natl. Acad. Sci. USA 2006, 103, 3816–3821. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W.; Dashkevicz, M.P.; Feighner, S.D. Lactobacilli and bile salt hydrolase in the murine intestinal tract. Appl. Environ. Microbiol. 1989, 55, 1848–1851. [Google Scholar] [PubMed]
- Stevens, C.E.; Hume, I.D. Comparative Physiology of the Vertebrate Digestive System, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Stackebrandt, E. The Family Gracilibacteraceae and Transfer of the Genus Lutispora into Gracilibacteraceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Germany, 2014. [Google Scholar]
- Alonso, B.L.; von Sierakowski, A.I.; Nieto, J.A.S.; Rosel, A.B. First report of human infection by Christensenalla minuta, a gram-negative, strickly anaerobic rod that inhabits the human intestine. Anaerobe 2017, 44, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Beerens, H.; Romond, C. Sulfate-reducing anaerobic bacteria in human feces. Am. J. Clin. Nutr. 1977, 30, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Dewhirst, F.E.; Fraser, G.J.; Paster, B.J.; Shames, B.; Murphy, J.C. Intracellular Campylobacter-like organism from ferrets and hamsters with proliferative bowel disease is a Desulfovibrio sp. J. Clin. Microbiol. 1994, 32, 1229–1237. [Google Scholar] [PubMed]
- Loubinoux, J.; Valente, F.M.A.; Pereira, I.A.; Costa, A.; Grimont, P.A.D.; Le Faou, A.E. Reclassification of the only species of the genus Desulfomonas, Desulfomonas pigra, as Desulfovibrio piger comb. Nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 1305–1308. [Google Scholar] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, K.H.; Kraus, J.; Dvorak, C.; Kilpper-Balz, R.; Collins, M.D.; Fischer, W. Transfer of Streptococcus lactis and related Streptococci to the genus Lactococcus gen. nov. Syst. Appl. Microbiol. 1985, 6, 183–195. [Google Scholar] [CrossRef]
- Maurice, C.F.; Knowles, S.C.; Ladau, J.; Pollard, K.S.; Fenton, A.; Pedersen, A.B.; Turnbaugh, P.J. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 2015, 9, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Diard, M.; Stecher, B.; Hardt, W.D. The streptomycin mouse model for Salmonella diarrhea: Functional analysis of the microbiota, the pathogen’s virulence factors, and the host’s mucosal immune response. Immunol. Rev. 2012, 245, 56–83. [Google Scholar] [CrossRef] [PubMed]
- Morowitz, M.J.; Carlisle, E.M.; Alverdy, J.C. Contributions of intestinal bacteria to nutrition and metabolism in the critically ill. Surg. Clin. 2011, 91, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Reeves, A.E.; Theriot, C.M.; Bergin, I.L.; Huffnagle, G.B.; Schloss, P.D.; Young, V.B. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2011, 2, 145–158. [Google Scholar] [CrossRef] [PubMed]
- DeBruyn, J.M.; Hauther, K.A. Postmortem succession of gut microbial communities in deceased human subjects. PeerJ 2017, 5, e3437. [Google Scholar] [CrossRef] [PubMed]
- Heimesaat, M.M.; Boelke, S.; Fischer, A.; Haag, L.-M.; Loddenkemper, C.; Kühl, A.A.; Göbel, U.B.; Bereswill, S. Comprehensive postmortem analyses of intestinal microbiota changes and bacterial translocation in human flora associated mice. PLoS ONE 2012, 7, e40758. [Google Scholar] [CrossRef] [PubMed]
- Marteau, P.; Pochart, P.; Doré, J.; Béra-Maillet, C.; Bernalier, A.; Corthier, G. Comparative study of bacterial groups within the human cecal and fecal microbiota. Appl. Environ. Microbiol. 2001, 67, 4939–4942. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet Group | n | Diet Provided | Duration of Exposure to Diet | Time in Captivity | |
|---|---|---|---|---|---|
| Sampled for feces | colony | 4 | Teklad 2018 rodent chow ad libitum | From birth | From birth |
| colony-transitional | 4 | 2 pieces Teklad 2018 rodent chow and greenhouse bulrush | 24 days | From birth | |
| wild-captured | 4 | Native bulrush, slices of carrot, sweet potato, apple, jicama, and Teklad 2018 rodent chow | 24–48 h | 24–48 h | |
| Sampled for foregut | colony | 3 | LabDiet 5326 rabbit chow ad libitum | From birth | From birth |
| wild | 3 | Native diet | From birth | n/a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allan, N.; Knotts, T.A.; Pesapane, R.; Ramsey, J.J.; Castle, S.; Clifford, D.; Foley, J. Conservation Implications of Shifting Gut Microbiomes in Captive-Reared Endangered Voles Intended for Reintroduction into the Wild. Microorganisms 2018, 6, 94. https://doi.org/10.3390/microorganisms6030094
Allan N, Knotts TA, Pesapane R, Ramsey JJ, Castle S, Clifford D, Foley J. Conservation Implications of Shifting Gut Microbiomes in Captive-Reared Endangered Voles Intended for Reintroduction into the Wild. Microorganisms. 2018; 6(3):94. https://doi.org/10.3390/microorganisms6030094
Chicago/Turabian StyleAllan, Nora, Trina A. Knotts, Risa Pesapane, Jon J. Ramsey, Stephanie Castle, Deana Clifford, and Janet Foley. 2018. "Conservation Implications of Shifting Gut Microbiomes in Captive-Reared Endangered Voles Intended for Reintroduction into the Wild" Microorganisms 6, no. 3: 94. https://doi.org/10.3390/microorganisms6030094
APA StyleAllan, N., Knotts, T. A., Pesapane, R., Ramsey, J. J., Castle, S., Clifford, D., & Foley, J. (2018). Conservation Implications of Shifting Gut Microbiomes in Captive-Reared Endangered Voles Intended for Reintroduction into the Wild. Microorganisms, 6(3), 94. https://doi.org/10.3390/microorganisms6030094