Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections



Abstract
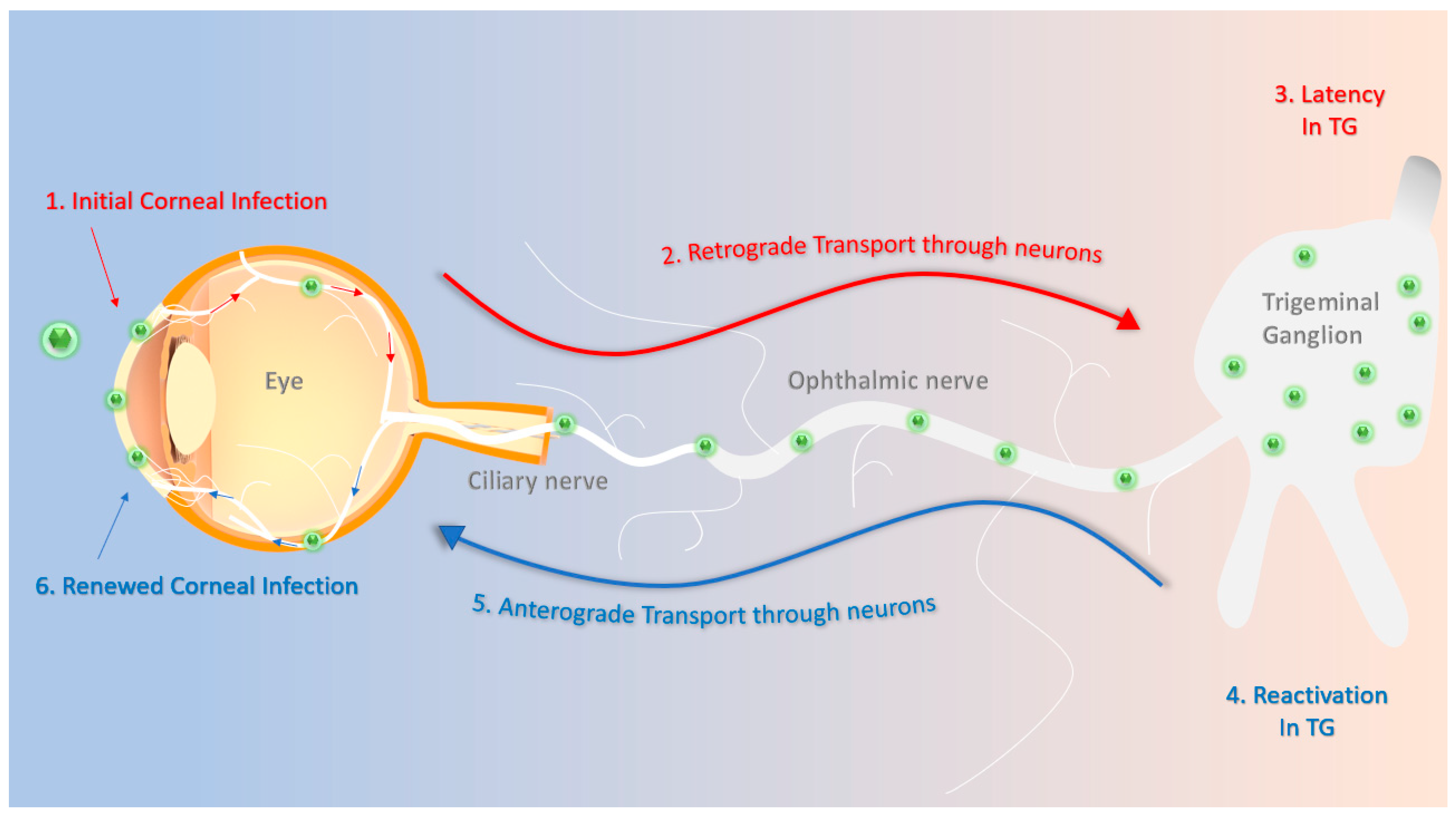
:1. Introduction
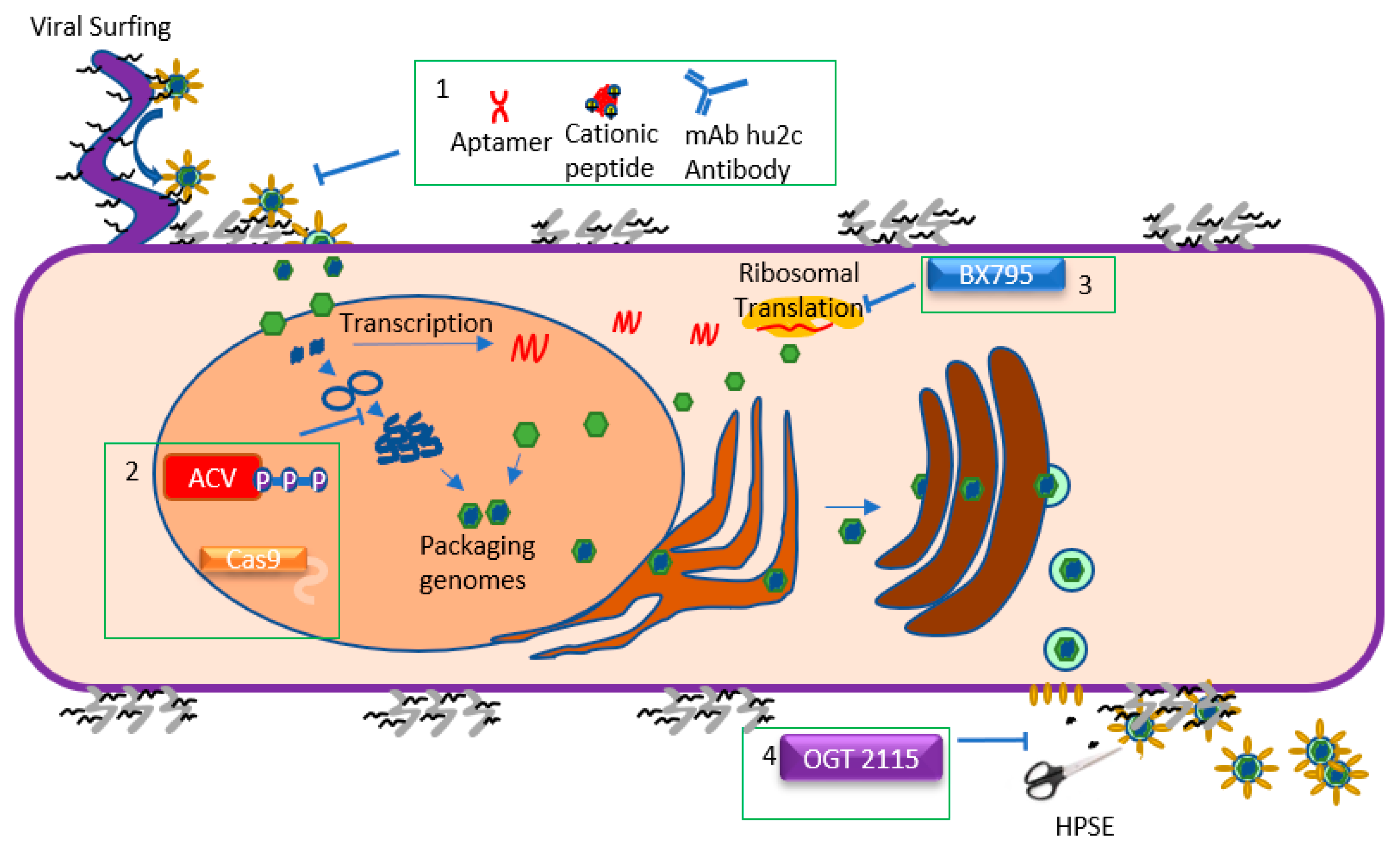
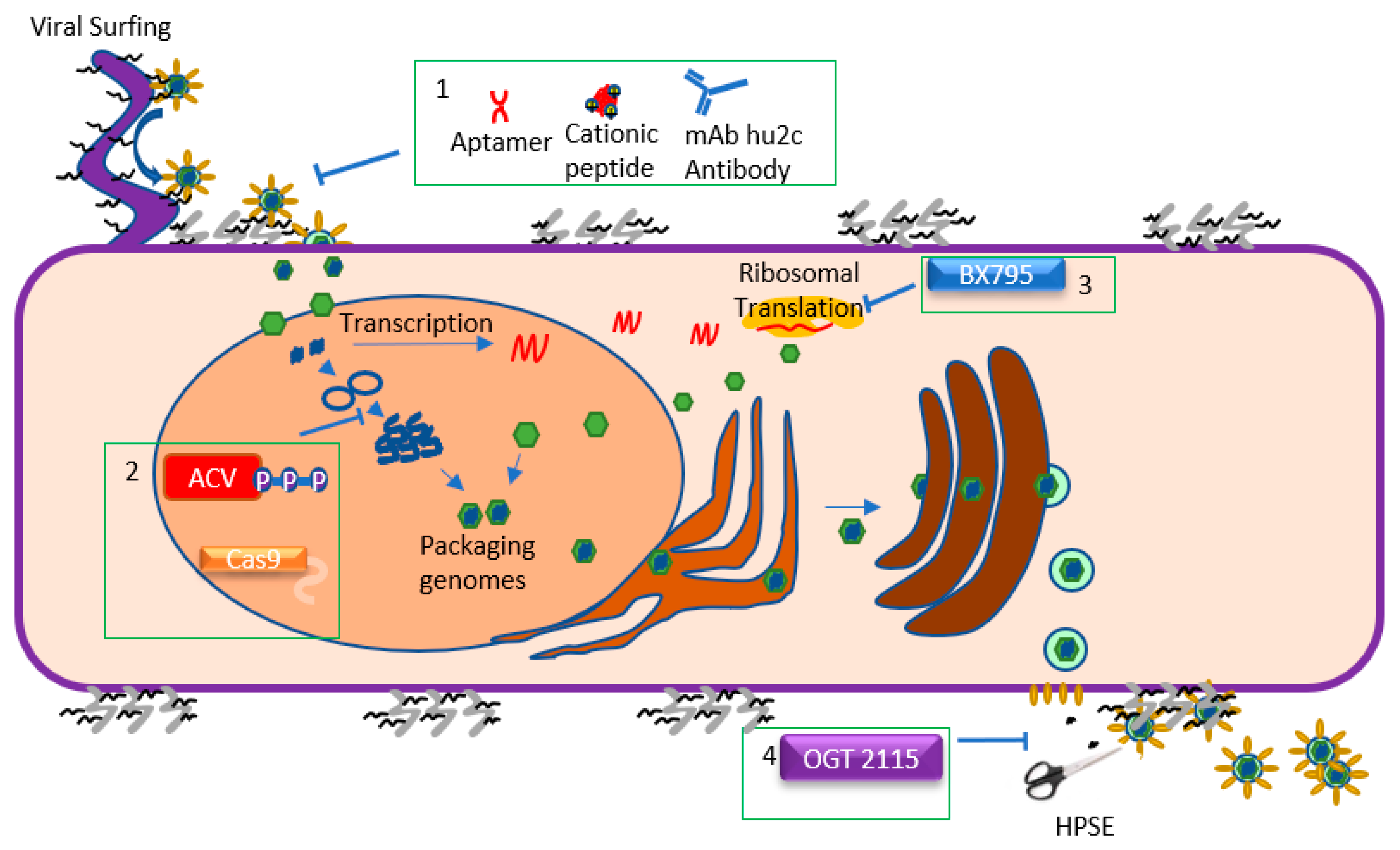
2. Viral Lifecycle: Attachment and Entry
3. Viral Lifecycle: Replication and Translation
4. Viral Lifecycle: Assembly and Egress
5. Challenges and Recent Progress in Controlling Ocular Herpes
6. Approved Therapy: Acyclovir and Other Nucleoside Analogs
7. Emerging Therapy: BX795
8. Emerging Therapy: Nucleic Acid Aptamers
9. Emerging Therapy: Cationic Peptide Therapies
10. Emerging Therapy: CRISPR/Cas9 System
11. Emerging Therapy: OGT 2115
12. Emerging Therapy: Antibodies
13. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liesegang, T.J. Herpes simplex virus epidemiology and ocular importance. Cornea 2001, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J. Herpesviruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. J. Am. Med. Assoc. 2006, 296, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Kasubi, M.J.; Nilsen, A.; Marsden, H.S.; Bergström, T.; Langeland, N.; Haarr, L. Prevalence of antibodies against herpes simplex virus types 1 and 2 in children and young people in an urban region in Tanzania. J. Clin. Microbiol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. Persistence in the population: epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; Chapter 36. [Google Scholar]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Koujah, L.; Suryawanshi, R.K.; Shukla, D. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell. Mol. Life Sci. 2019, 76, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.V.; Shukla, D. Corneal latency and transmission of herpes simplex virus-1. Future Virol. 2011, 6, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef] [PubMed]
- Lobo, A.M.; Agelidis, A.M.; Shukla, D. Pathogenesis of herpes simplex keratitis: The host cell response and ocular surface sequelae to infection and inflammation. Ocul. Surf. 2019, 17, 40–49. [Google Scholar] [CrossRef]
- Liesegang, T.J.; Melton, L.J., III; Daly, P.J.; Ilstrup, D.M. Epidemiology of Ocular Herpes Simplex: Incidence in Rochester, Minn, 1950 Through 1982. Arch. Ophthalmol. 1989, 107, 1155–1159. [Google Scholar] [CrossRef]
- Remeijer, L.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Human herpes simplex virus keratitis: The pathogenesis revisited. Ocul. Immunol. Inflamm. 2004, 12, 255–285. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Farooq, A.V.; Valyi-Nagy, T.; Shukla, D. Mediators and Mechanisms of Herpes Simplex Virus Entry into Ocular Cells. Curr. Eye Res. 2010, 35, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, U.; Wang, S.; Tormanen, K.; Matundan, H.; Ljubimov, A.V.; Ghiasi, H. Role of Herpes Simplex Virus Type 1 (HSV-1) Glycoprotein K (gK) Pathogenic CD8+ T Cells in Exacerbation of Eye Disease. Front. Immunol. 2018, 9, 2895. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Invest. 2001, 108, 503–510. [Google Scholar] [CrossRef]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Tiwari, V.; Clement, C.; Xu, D.; Valyi-Nagy, T.; Yue, B.Y.J.T.; Liu, J.; Shukla, D. Role for 3-O-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J. Virol. 2006, 80, 8970–8980. [Google Scholar] [CrossRef]
- Shah, A.; Farooq, A.V.; Tiwari, V.; Kim, M.-J.; Shukla, D. HSV-1 infection of human corneal epithelial cells: Receptormediated entry and trends of re-infection. Mol. Vision 2010, 16, 2476–2486. [Google Scholar]
- Akhtar, J.; Kovacs, M.; Oh, M.; Tiwari, V.; Jani, A.; Shukla, D. Novel Aspects of Herpes Simplex Virus 1 Entry Into Human Conjunctival Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5517. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated Transport of Incoming Herpes Simplex Virus 1 Capsids to the Nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S.N. Human herpes simplex virus: Life cycle and development of inhibitors. Biochemistry 2014, 79, 1635–1652. [Google Scholar] [CrossRef] [PubMed]
- Zaichick, S.; Bohannon, K.; Hughes, A.; Sollars, P.; Pickard, G.; Smith, G. The Herpesvirus VP1/2 Protein Is an Effector of Dynein-Mediated Capsid Transport and Neuroinvasion. Cell Host Microbe 2013, 13, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, A.M.; Newcomb, W.W.; Brown, J.C. Herpes simplex virus replication: Roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 2009, 83, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Bernad, R.; van der Velde, H.; Fornerod, M.; Pickersgill, H. Nup358/RanBP2 Attaches to the Nuclear Pore Complex via Association with Nup88 and Nup214/CAN and Plays a Supporting Role in CRM1-Mediated Nuclear Protein Export. Mol. Cell. Biol. 2004, 24, 2373–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri-Goldin, R.M.; Goldin, A.L.; Holland, L.E.; Glorioso, J.C.; Levine, M. A Expression of Herpes Simplex Virus and y Genes Integrated in Mammalian Cells and Their Induction by an α Gene Product. Mol. Cell. Biol. 1983, 3, 2028–2044. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Proença, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Boutell, C.; Davido, D.J. HSV-1 ICP0: paving the way for viral replication. Future Virol. 2011, 6, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected Cell Protein (ICP)47 Enhances Herpes Simplex Virus Neurovirulence by Blocking the CD8+ T Cell Response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Pesola, J.M.; Zhu, J.; Knipe, D.M.; Coen, D.M. Herpes Simplex Virus 1 Immediate-Early and Early Gene Expression during Reactivation from Latency under Conditions That Prevent Infectious Virus Production. J. Virol. 2006, 80, 6196. [Google Scholar] [CrossRef]
- Boehmer, P.E.; Lehman, I.R. Herpes simplex virus DNA replication. Annu. Rev. Biochem. 1997, 66, 347–384. [Google Scholar] [CrossRef] [PubMed]
- Mingo, R.M.; Han, J.; Newcomb, W.W.; Brown, J.C. Replication of herpes simplex virus: Egress of progeny virus at specialized cell membrane sites. J. Virol. 2012, 86, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef] [PubMed]
- Sedlackova, L.; Stephen, A. Rice Herpes Simplex Virus Type 1 Immediate-Early Protein ICP27 Is Required for Efficient Incorporation of ICP0 and ICP4 into Virions. Journal of Virology 2008, 82, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C. Herpesvirus Assembly and Egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson-Holley, M.; Baines, J.; Roller, R.; Knipe, D.M. Herpes simplex virus 1 UL31 and UL34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 2004, 78, 5591–5600. [Google Scholar] [CrossRef] [PubMed]
- Bond, V.C.; Person, S. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 1984, 132, 368–376. [Google Scholar] [CrossRef]
- Hutchinson, L.; Goldsmith, K.; Snoddy, D.; Ghosh, H.; Graham, F.L.; Johnson, D.C. Identification and Characterization of a Novel Herpes Simplex Virus Glycoprotein, gK, Involved in Cell Fusion. J. Virol. 1992, 66, 5603–5609. [Google Scholar]
- Kim, I.J.; Chouljenko, V.N.; Walker, J.D.; Kousoulas, K.G. Herpes Simplex Virus 1 Glycoprotein M and the Membrane-Associated Protein UL11 Are Required for Virus-Induced Cell Fusion and Efficient Virus Entry. J. Virol. 2013, 87, 8029–8037. [Google Scholar] [CrossRef] [Green Version]
- Skepper, J.N.; Whiteley, A.; Browne, H.; Minson, A. Herpes Simplex Virus Nucleocapsids Mature to Progeny Virions by an Envelopment → Deenvelopment → Reenvelopment Pathway. J. Virol. 2001, 75, 5697–5702. [Google Scholar] [CrossRef]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.L.; Baines, J.D. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J. Virol. 2010, 84, 9889–9896. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhou, S.; Gao, S.; Deng, H. Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 2019, 10, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef]
- Dargan, D.J.; Subak-Sharpe, J.H. The effect of herpes simplex virus type 1 L-particles on virus entry, replication, and the infectivity of naked herpesvirus DNA. Virology 1997, 239, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef]
- Agelidis, A.M.; Hadigal, S.R.; Jaishankar, D.; Shukla, D. Viral Activation of Heparanase Drives Pathogenesis of Herpes Simplex Virus-1. Cell Rep. 2017, 20, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, J.; Yadavalli, T.; Agelidis, A.M.; Shukla, D. Host Enzymes Heparanase and Cathepsin L Promote Herpes Simplex Virus 2 Release from Cells. J. Virol. 2018, 92, e01179-18. [Google Scholar] [CrossRef]
- Coleman, J.L.; Shukla, D. Recent advances in vaccine development for herpes simplex virus types i and II. Hum. Vaccines Immunother. 2013, 9, 729–735. [Google Scholar] [CrossRef]
- Wilhelmus, K.R. The treatment of herpes simplex virus epithelial keratitis. Trans. Am. Ophthalmol. Soc. 2000, 98, 505–532. [Google Scholar] [PubMed]
- Kimberlin, D.W.; Whitley, R.J. Antiviral therapy of HSV-1 and -2. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; Chapter 64. [Google Scholar]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.S.; Fakioglu, E.; Herold, B.C. Novel approaches in fighting herpes simplex virus infections. Expert Rev. Anti-Infect. Ther. 2009, 7, 559–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsatsos, M.; MacGregor, C.; Athanasiadis, I.; Moschos, M.M.; Hossain, P.; Anderson, D. Herpes simplex virus keratitis: an update of the pathogenesis and current treatment with oral and topical antiviral agents. Clin. Exp. Ophthalmol. 2016, 44, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Phulke, S.; Kaushik, S.; Kaur, S.; Pandav, S. Steroid-induced Glaucoma: An Avoidable Irreversible Blindness. J. Curr. Glaucoma Pract. 2017, 11, 67–72. [Google Scholar] [PubMed]
- Yildiz, C.; Ozsurekci, Y.; Gucer, S.; Cengiz, A.; Topaloglu, R. Acute kidney injury due to acyclovir. CEN Case Rep. 2013, 2, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, R.; Johnson, M. Acyclovir Nephrotoxicity: A Case Report Highlighting the Importance of Prevention, Detection, and Treatment of Acyclovir-Induced Nephropathy. Case Rep. Med. 2010, 2010, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.L.; Chodosh, J. Herpes Simplex Virus Keratitis: A Treatment Guideline. Available online: http://one.aao.org/clinical-statement/herpes-simplex-virus-keratitis-treatment-guideline (accessed on 1 October 2019).
- Whitley, R.J.; Gnann, J.W. Acyclovir: A Decade Later. N. Engl. J. Med. 1992, 327, 782–789. [Google Scholar] [CrossRef]
- Elion, G.B. Acyclovir: Discovery, mechanism of action, and selectivity. J. Med Virol. 1993, 41, 2–6. [Google Scholar] [CrossRef]
- Elion, G.B. Mechanism of action and selectivity of acyclovir. Am. J. Med. 1982, 73, 7–13. [Google Scholar] [CrossRef]
- Colin, J.; Hoh, H.B.; Easty, D.L.; Herbort, C.P.; Resnikoff, S.; Rigal, D.; Romdane, K. Ganciclovir ophthalmic gel (Virgan; 0.15%) in the treatment of herpes simplex keratitis. Cornea 1997, 16, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.Y.; Hong, B.Y. Ganciclovir ophthalmic gel 0.15% for the treatment of acute herpetic keratitis: background, effectiveness, tolerability, safety, and future applications. Ther. Clin. Risk Manag. 2014, 10, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Beutner, K.R.; Friedman, D.J.; Forszpaniak, C.; Andersen, P.L.; Wood, M.J. Valaciclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults. Antimicrob. Agents Chemother. 1995, 39, 1546–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyring, S.K.; Baker, D.; Snowden, W. Valacyclovir for herpes simplex virus infection: long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J. Infect. Dis. 2002, 186 (Suppl. 1), 40. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.; Engst, R.; Corriveau, C.; Robillard, N.; Trottier, S.; Van Slycken, S.; Crann, R.A.; Locke, L.A.; Saltzman, R.; Palestine, A.G. Famciclovir for ophthalmic zoster: A randomised aciclovir controlled study. Br. J. Ophthalmol. 2001, 85, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Yaldiz, M.; Solak, B.; Kara, R.O.; Cosansu, N.; Erdem, M.T. Comparison of Famciclovir, Valaciclovir, and Brivudine Treatments in Adult Immunocompetent Patients With Herpes Zoster. Am. J. Ther. 2016, 25, e626–e634. [Google Scholar] [CrossRef] [PubMed]
- Sudesh, S.; Laibson, P.R. The impact of the herpetic eye disease studies on the management of herpes simplex virus ocular infections. Curr. Opin. Ophthalmol. 1999, 10, 230–233. [Google Scholar] [CrossRef]
- Wilhelmus, K.R.; Gee, L.; Hauck, W.W.; Kurinij, N.; Dawson, C.R.; Jones, D.B.; Barron, B.A.; Kaufman, H.E.; Sugar, J.; Hyndiuk, R.A. Herpetic Eye Disease Study. A controlled trial of topical corticosteroids for herpes simplex stromal keratitis. Ophthalmology 1994, 101, 188–1896. [Google Scholar]
- Elion, G.B.; Furman, P.A.; Fyfe, J.A.; Miranda, P.d.; Beauchamp, L.; Schaeffer, H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl)guanine. Proc. Natl. Acad. Sci. USA 1977, 74, 5716–5720. [Google Scholar] [CrossRef] [Green Version]
- Beck, R.W.; Asbell, P.A.; Cohen, E.J.; Dawson, C.R.; Hyndiuk, R.A.; Jones, D.B.; Kaufman, H.E.; Kip, K.E.; Kurinij, N.; Moke, P.S.; et al. Oral acyclovir for herpes simplex virus eye disease: Effect on prevention of epithelial keratitis and stromal keratitis. Arch. Ophthalmol. 2000, 118, 1030–1036. [Google Scholar]
- Gnann, J.W., Jr.; Barton, N.H.; Whitley, R.J. Acyclovir: Mechanism of Action, Pharmacokinetics, Safety and Clinical Applications. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1983, 3, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lass, J.H.; Langston, R.H.S.; Stephen Foster, C.; Pavan-Langston, D. Antiviral medications and corneal wound healing. Antivir. Res. 1984, 4, 143–157. [Google Scholar] [CrossRef]
- McLaren, C.; Corey, L.; Dekket, C.; Barry, D.W. In vitro sensitivity to acyclovir in genital herpes simplex viruses from acyclovir-treated patients. J. Infect. Dis. 1983, 148, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.A.; Keller, P.M.; Furman, P.A.; Miller, R.L.; Elion, G.B. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 1978, 253, 8721–8727. [Google Scholar] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.H.; Saral, R.; Santos, G.W.; Laskin, O.L.; Lietman, P.S.; McLaren, C.; Barry, D.W. Isolation and characterisation of resistant Herpes simplex virus after acyclovir therapy. Lancet 1982, 1, 421–423. [Google Scholar] [CrossRef]
- Jiang, Y.C.; Feng, H.; Lin, Y.C.; Guo, X.R. New strategies against drug resistance to herpes simplex virus. Int. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S.; Schnipper, L.E.; Marlowe, S.I.; Kowalsky, P.N.; Hershey, B.J.; Levin, M.J. Resistance to Antiviral Drugs of Herpes Simplex Virus Isolated from a Patient Treated with Acyclovir. N. Engl. J. Med. 2010, 306, 343–346. [Google Scholar] [CrossRef]
- Larder, B.A.; Cheng, Y.; Darby, G. Characterization of Abnormal Thymidine Kinases Induced by Drug-resistant Strains of Herpes Simplex Virus Type 1. J. Gen. Virol. 1983, 64, 523–532. [Google Scholar] [CrossRef]
- Yadavalli, T.; Ames, J.; Agelidis, A.; Suryawanshi, R.; Jaishankar, D.; Hopkins, J.; Thakkar, N.; Koujah, L.; Shukla, D. Drug-encapsulated carbon (DECON): A novel platform for enhanced drug delivery. Sci. Adv. 2019, 5, eaax0780. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Plater, L.; Peggie, M.; Cohen, P. Use of the Pharmacological Inhibitor BX795 to Study the Regulation and Physiological Roles of TBK1 and IκB Kinase ϵ. J. Biol. Chem. 2009, 284, 14136–14146. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, D.; Yakoub, A.M.; Yadavalli, T.; Agelidis, A.; Thakkar, N.; Hadigal, S.; Ames, J.; Shukla, D. An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Cheshenko, N.; Trepanier, J.B.; Stefanidou, M.; Buckley, N.; Gonzalez, P.; Jacobs, W.; Herold, B.C. HSV activates Akt to trigger calcium release and promote viral entry: Novel candidate target for treatment and suppression. FASEB J. 2013, 27, 2584–2599. [Google Scholar] [CrossRef] [PubMed]
- Chuluunbaatar, U.; Roller, R.; Feldman, M.E.; Brown, S.; Shokat, K.M.; Mohr, I. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 2010, 24, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.C.B.; Hayashi, K.; Kumar, P.K.R. Aptamer that binds to the gD protein of herpes simplex virus 1 and efficiently inhibits viral entry. J. Virol. 2012, 86, 6732–6744. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Escudero-Abarca, B.I.; Suh, S.H.; Jaykus, L.-A. Generation and characterization of nucleic acid aptamers targeting the capsid P domain of a human norovirus GII.4 strain. J. Biotechnol. 2015, 209, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadavalli, T.; Agelidis, A.; Jaishankar, D.; Mangano, K.; Thakkar, N.; Penmetcha, K.; Shukla, D. Targeting Herpes Simplex Virus-1 gD by a DNA Aptamer Can Be an Effective New Strategy to Curb Viral Infection. Mol. Ther. Nucleic Acids 2017, 9, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultmann, H.; Busse, J.S.; Brandt, C.R. Modified FGF4 signal peptide inhibits entry of herpes simplex virus type 1. J. Virol. 2001, 75, 2634–2645. [Google Scholar] [CrossRef] [PubMed]
- Bultmann, H.; Girdaukas, G.; Kwon, G.S.; Brandt, C.R. The virucidal EB peptide protects host cells from herpes simplex virus type 1 infection in the presence of serum albumin and aggregates proteins in a detergent-like manner. Antimicrob. Agents Chemother. 2010, 54, 4275–4289. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Liu, J.; Valyi-Nagy, T.; Shukla, D. Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J. Biol. Chem. 2011, 286, 25406–25415. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J.; Antoine, T.E.; Farooq, A.V.; Valyi-Nagy, T.; Shukla, D. An Investigative Peptide–Acyclovir Combination to Control Herpes Simplex Virus Type 1 Ocular Infection. Invest. Ophthalmol. Vis. Sci. 2013, 54, 6373–6381. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, D.; Buhrman, J.S.; Valyi-Nagy, T.; Gemeinhart, R.A.; Shukla, D. Extended Release of an Anti–Heparan Sulfate Peptide From a Contact Lens Suppresses Corneal Herpes Simplex Virus-1 Infection. Invest. Ophthalmol. Vis. Sci. 2016, 57, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.R.; Akkarawongsa, R.; Altmann, S.; Jose, G.; Kolb, A.W.; Waring, A.J.; Lehrer, R.I. Evaluation of a theta-defensin in a Murine model of herpes simplex virus type 1 keratitis. Invest. Ophthalmol. Vis. Sci. 2007, 48, 5118–5124. [Google Scholar] [CrossRef]
- Jose, G.G.; Larsen, I.V.; Gauger, J.; Carballo, E.; Stern, R.; Brummel, R.; Brandt, C.R. A cationic peptide, TAT-Cd°, inhibits herpes simplex virus type 1 ocular infection in vivo. Invest. Ophthalmol. Vis. Sci. 2013, 54, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Bultmann, H.; Teuton, J.; Brandt, C.R. Addition of a C-terminal cysteine improves the anti-herpes simplex virus activity of a peptide containing the human immunodeficiency virus type 1 TAT protein transduction domain. Antimicrob. Agents Chemother. 2007, 51, 1596–1607. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev. Med. Virol. 2014, 24, 186–218. [Google Scholar] [CrossRef]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Lander, E.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, S.H.; Doudna, J.A. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol. Cell 2015, 58, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.G.; Bruggeling, C.E.; Schürch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Sun, L.; Gao, D.; Ding, C.; Li, Z.; Li, Y.; Cun, W.; Li, Q. High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases. PLoS Pathog. 2014, 10, e1004090. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stephen, R. Quake RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA 2014, 111, 13157–13162. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.A.; Stefanovic, T.; Tscharke, D.C. Engineering herpes simplex viruses by infection-transfection methods including recombination site targeting by CRISPR/Cas9 nucleases. J. Virol. Methods 2015, 213, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, X.; Peng, X.; Xiang, Y.; Song, S.; Wang, Y.; Chen, L.; Xin, V.W.; Lyu, Y.; Ji, J.; et al. CRISPR/Cas9 genome editing technology significantly accelerated herpes simplex virus research. Cancer Gene Ther. 2018, 25, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Roehm, P.C.; Shekarabi, M.; Wollebo, H.S.; Bellizzi, A.; He, L.; Salkind, J.; Khalili, K. Inhibition of HSV-1 Replication by Gene Editing Strategy. Sci. Rep. 2016, 6, 23146. [Google Scholar] [CrossRef] [PubMed]
- Courtney, S.M.; Hay, P.A.; Buck, R.T.; Colville, C.S.; Phillips, D.J.; Scopes, D.I.C.; Pollard, F.C.; Page, M.J.; Bennett, J.M.; Hircock, M.L.; et al. Furanyl-1,3-thiazol-2-yl and benzoxazol-5-yl acetic acid derivatives: novel classes of heparanase inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 2295–2299. [Google Scholar] [CrossRef]
- Krawczyk, A.; Arndt, M.A.E.; Grosse-Hovest, L.; Weichert, W.; Giebel, B.; Dittmer, U.; Hengel, H.; Jäger, D.; Schneweis, K.E.; Eis-Hübinger, A.M.; et al. Overcoming drug-resistant herpes simplex virus (HSV) infection by a humanized antibody. Proc. Natl. Acad. Sci. USA 2013, 110, 6760–6765. [Google Scholar] [CrossRef] [Green Version]
- Hadigal, S.; Shukla, D. Exploiting herpes simplex virus entry for novel therapeutics. Viruses 2013, 5, 1447–1465. [Google Scholar] [CrossRef]
- Antoine, T.E.; Park, P.J.; Shukla, D. Glycoprotein targeted therapeutics: A new era of anti-herpes simplex virus-1 therapeutics. Rev. Med. Virol. 2013, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Alt, M.; Dirks, M.; Buch, A.; Heilingloh, C.S.; Dittmer, U.; Giebel, B.; Görgens, A.; Palapys, V.; Kasper, M.; et al. A Therapeutic Antiviral Antibody Inhibits the Anterograde Directed Neuron-to-Cell Spread of Herpes Simplex Virus and Protects against Ocular Disease. Front. Microbiol. 2017, 8, 2115. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Name | Mechanism of Action | Application | Recommended Dosage | Reference |
|---|---|---|---|---|
| Acyclovir | Guanosine Analog | Systemic | 400 mg 3-5 times/day | [62,63,64] |
| Ganciclovir | Guanosine Analog | Topical | 1 drop of 0.15% gel 5 times/day | [65,66] |
| Valacyclovir | Guanosine Analog | Systemic | 500 mg 2 times/day | [67,68] |
| Famciclovir | Guanosine analog | Systemic | 250 mg 2 times/day | [61,69,70] |
| Trifluridine | Thymine Analog | Topical | 1 drop of solution 9 times/day | [61,71] |
| Corticosteroids | Anti-inflammatory agent | Systemic, topical | Frequency based on severity of Inflammation | [72] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koganti, R.; Yadavalli, T.; Shukla, D. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms 2019, 7, 429. https://doi.org/10.3390/microorganisms7100429
Koganti R, Yadavalli T, Shukla D. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms. 2019; 7(10):429. https://doi.org/10.3390/microorganisms7100429
Chicago/Turabian StyleKoganti, Raghuram, Tejabhiram Yadavalli, and Deepak Shukla. 2019. "Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections" Microorganisms 7, no. 10: 429. https://doi.org/10.3390/microorganisms7100429
APA StyleKoganti, R., Yadavalli, T., & Shukla, D. (2019). Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms, 7(10), 429. https://doi.org/10.3390/microorganisms7100429