Structural and Functional Dynamics of Staphylococcus aureus Biofilms and Biofilm Matrix Proteins on Different Clinical Materials

 , , ,
, , ,
Abstract
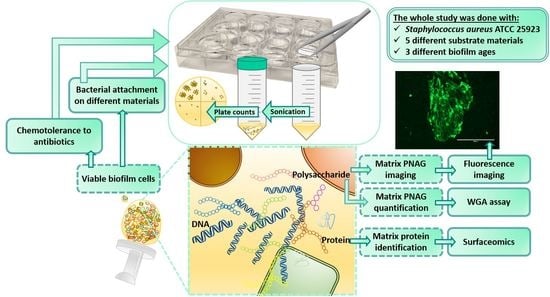
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Atomic Force Microscopy (AFM)
2.3. Bacterial Culture and Biofilm Formation
2.4. Quantification of Biofilm Formation on Different Materials
2.5. Quantification of Matrix-Associated Poly-N-Acetyl-β-(1-6)-Glucosamine
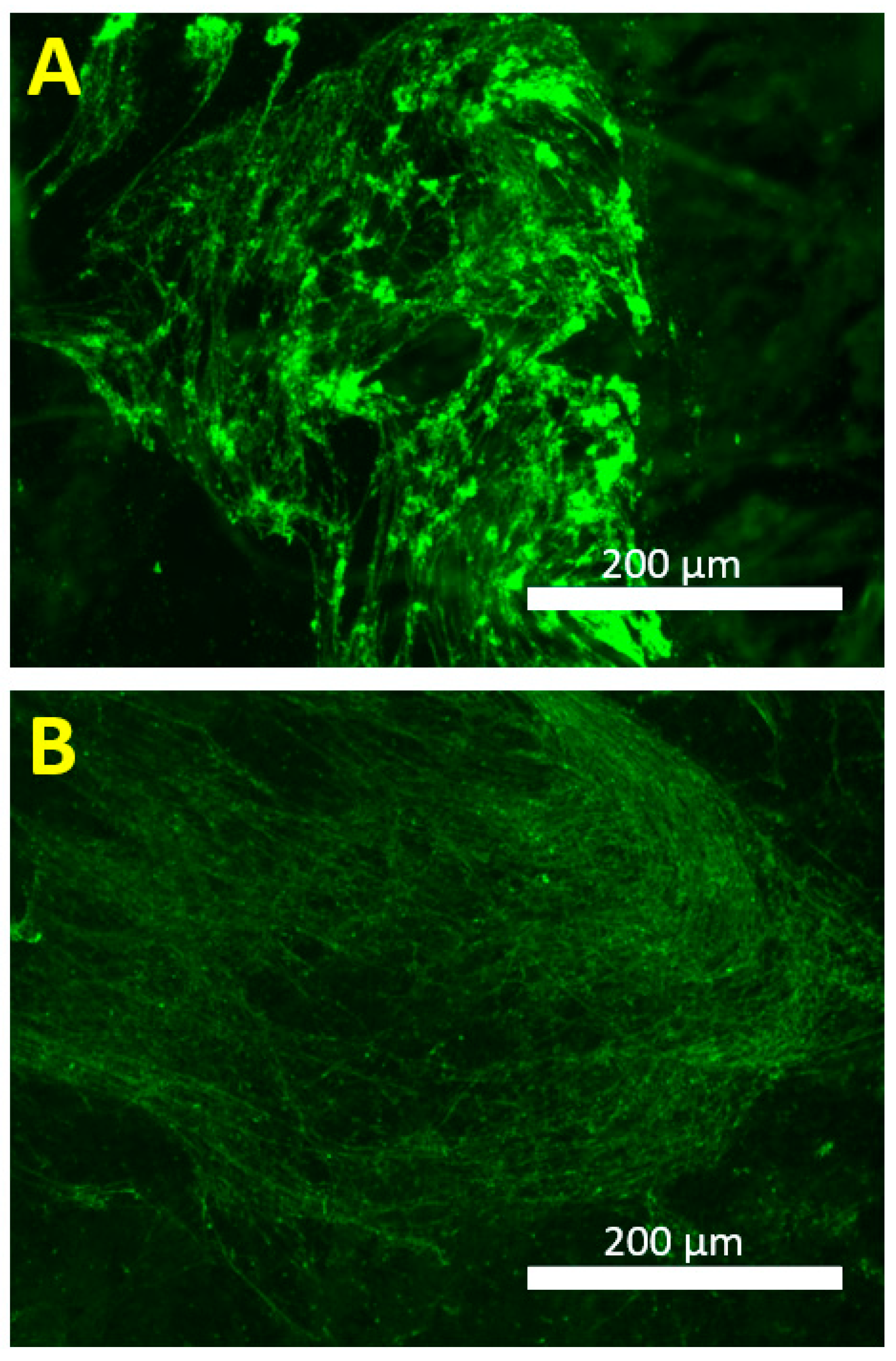
2.6. Fluorescence Imaging
2.7. Trypsin Shaving of Matrix-Associated Proteins
2.8. Identification of Trypsin-Released Proteins/Peptides by LC–MS/MS
2.9. Chemotolerance Assays
2.10. Data Processing and Statistical Analysis
3. Results
3.1. HA and PG Exhibited the Largest Surface Roughness
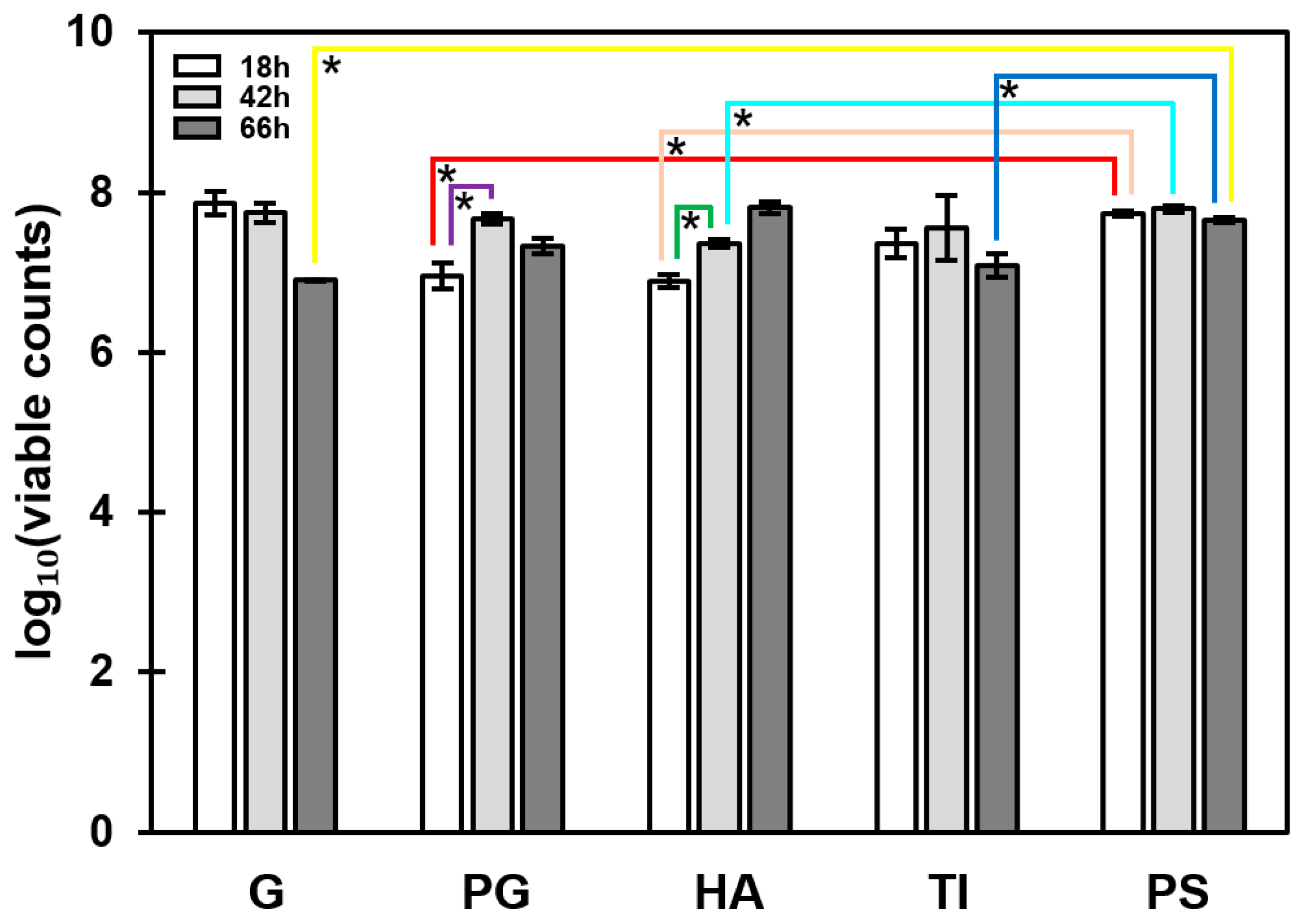
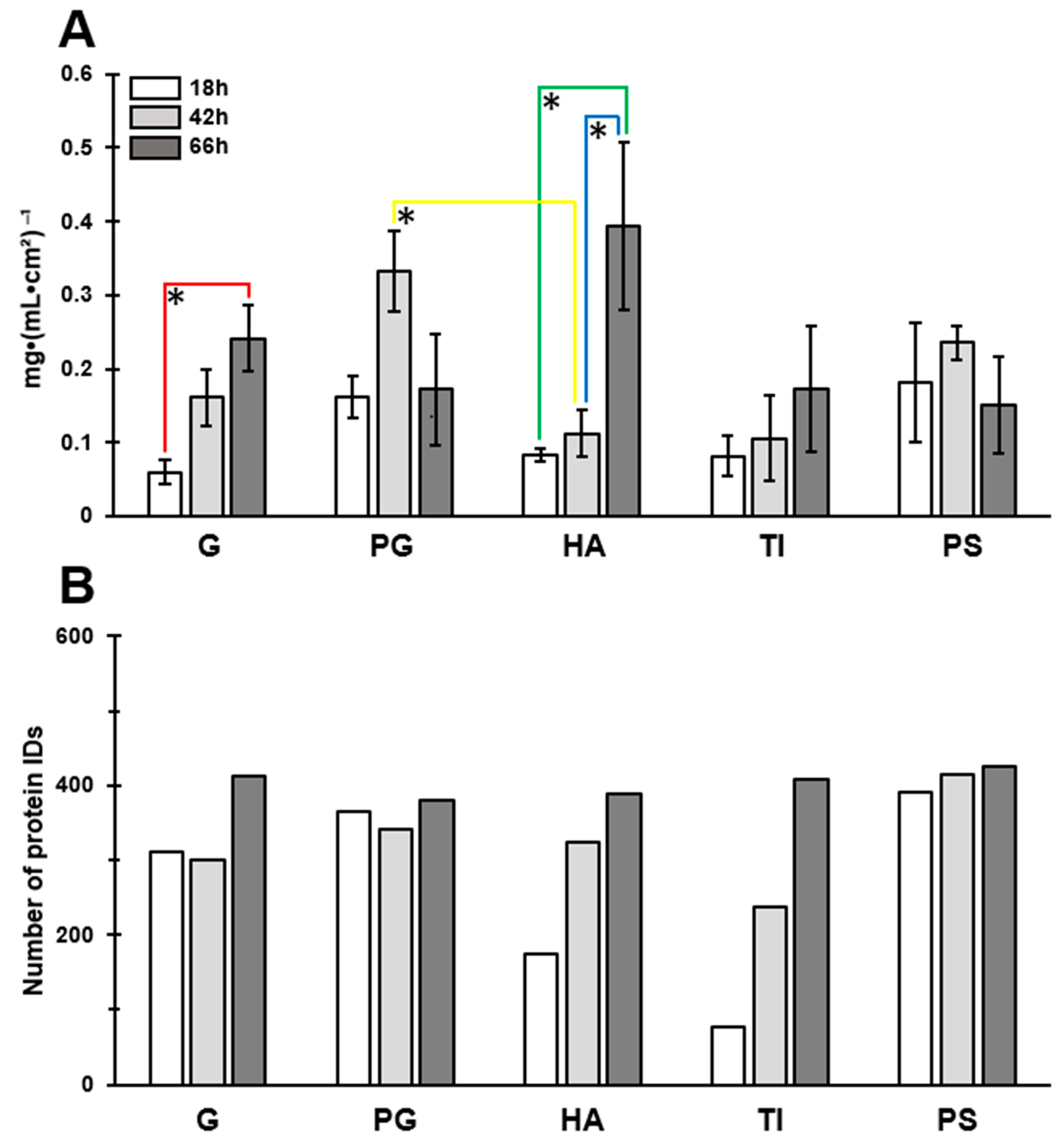
3.2. The Most Significant Time-Dependent Increase in Biofilm Formation Was Detected on HA
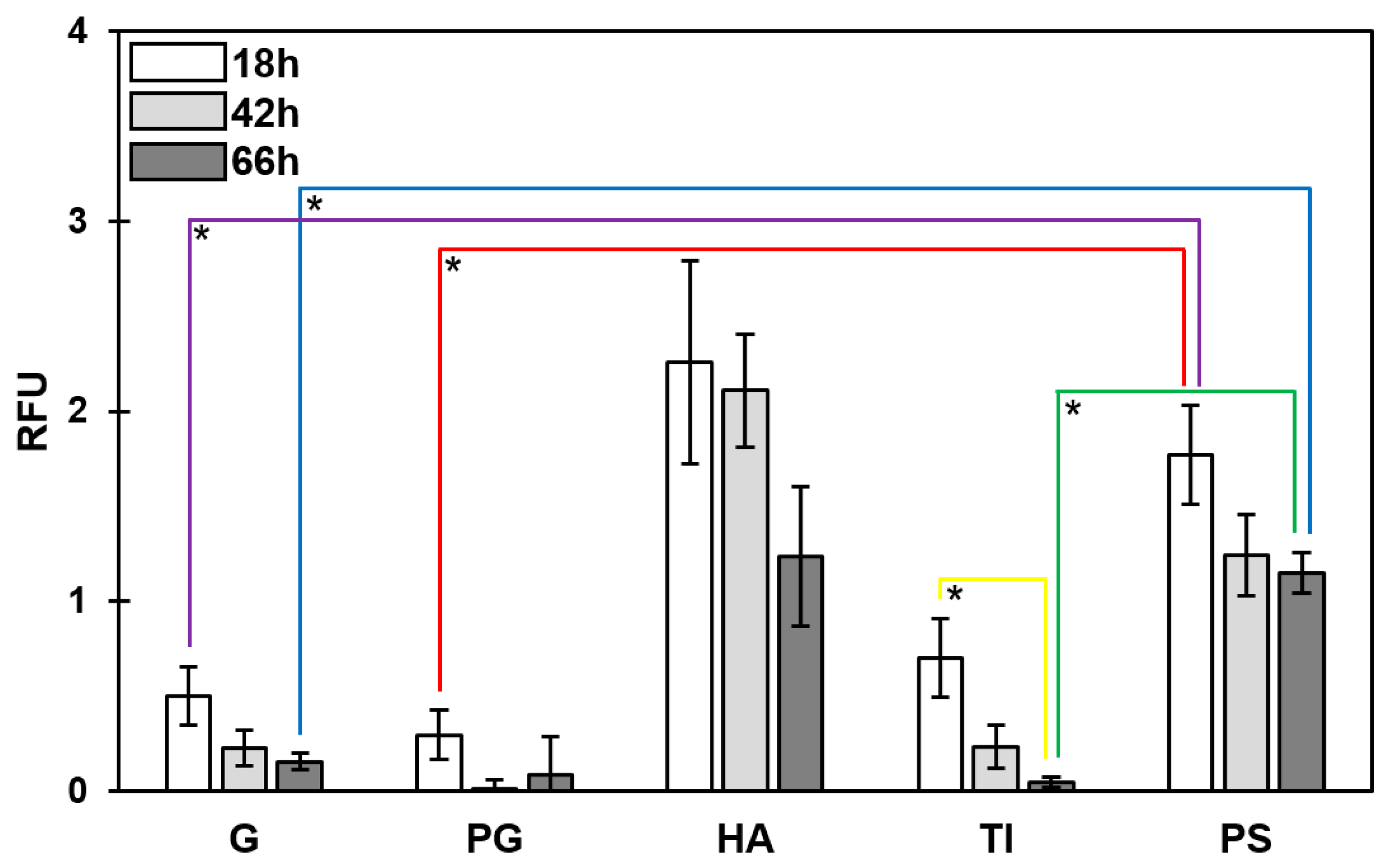
3.3. Temporal Decrease in the Total PNAG Amount Was Detected in All Biofilms
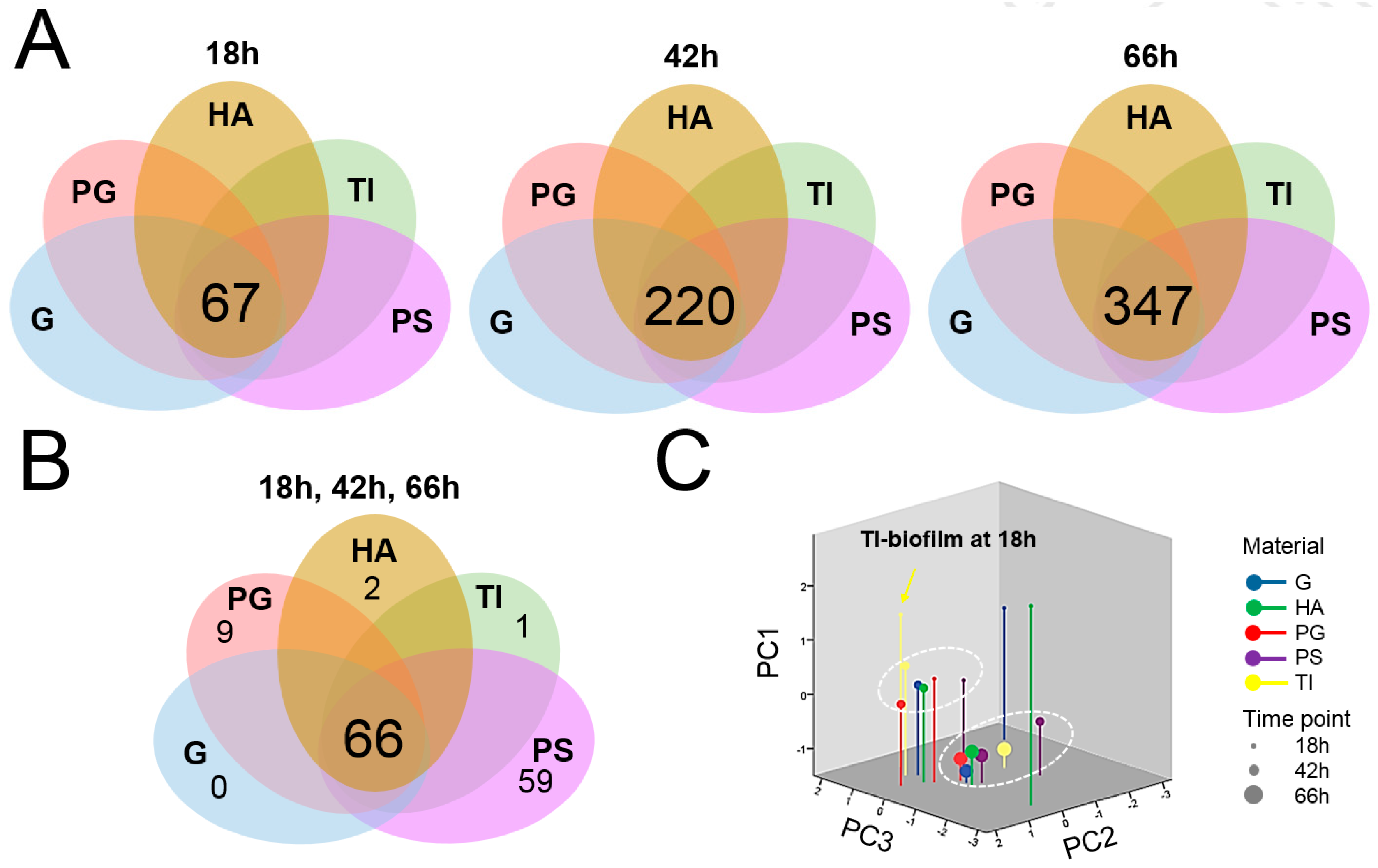
3.4. A Total of 66 Proteins Were Shared by All Biofilms
3.5. Protein Moonlighters Formed the Largest Fraction of the Core Surfaceome
3.6. Greatest Time-Dependent Variations Were Observed for TI- and HA-Associated Surfaceomes
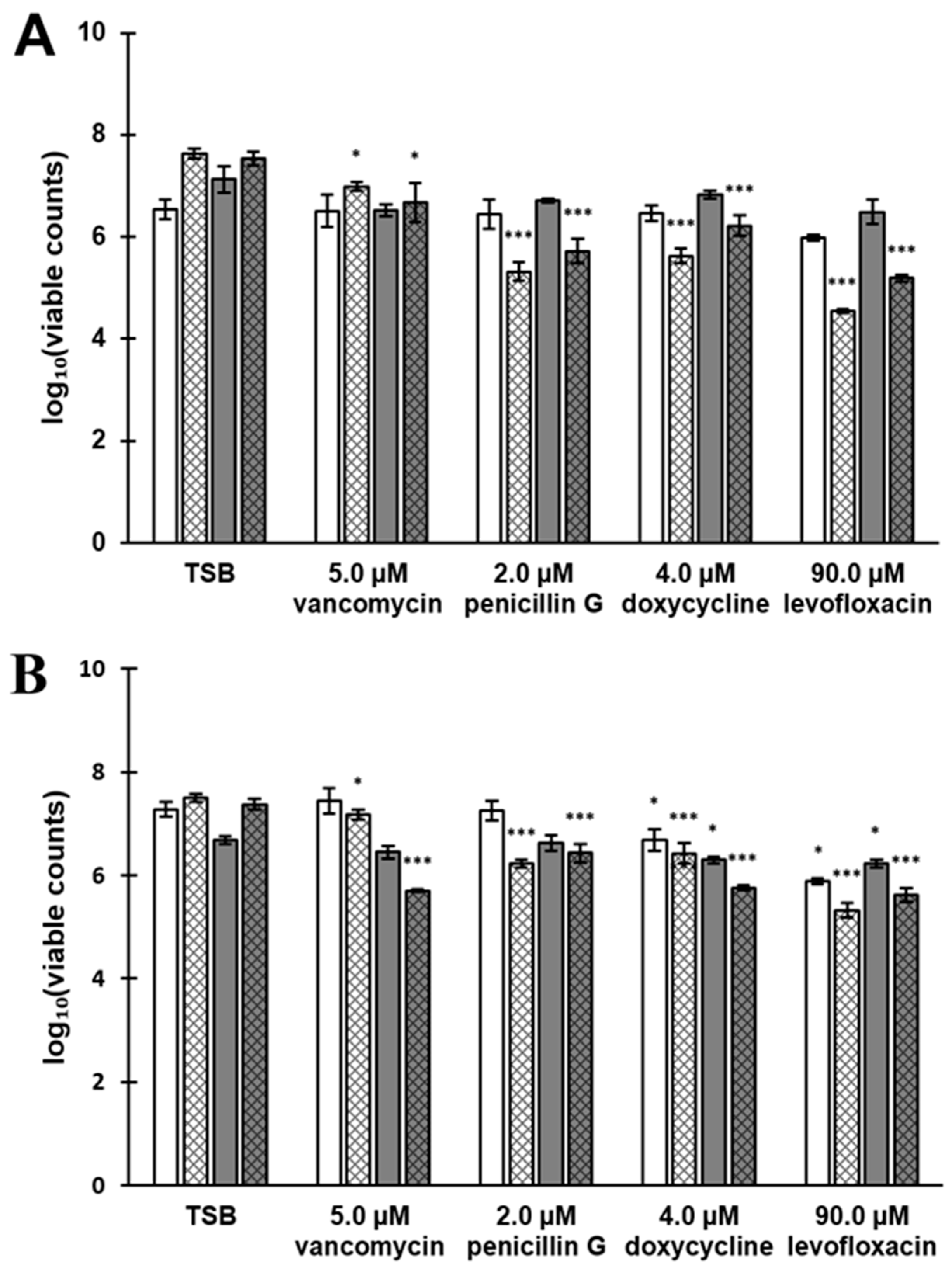
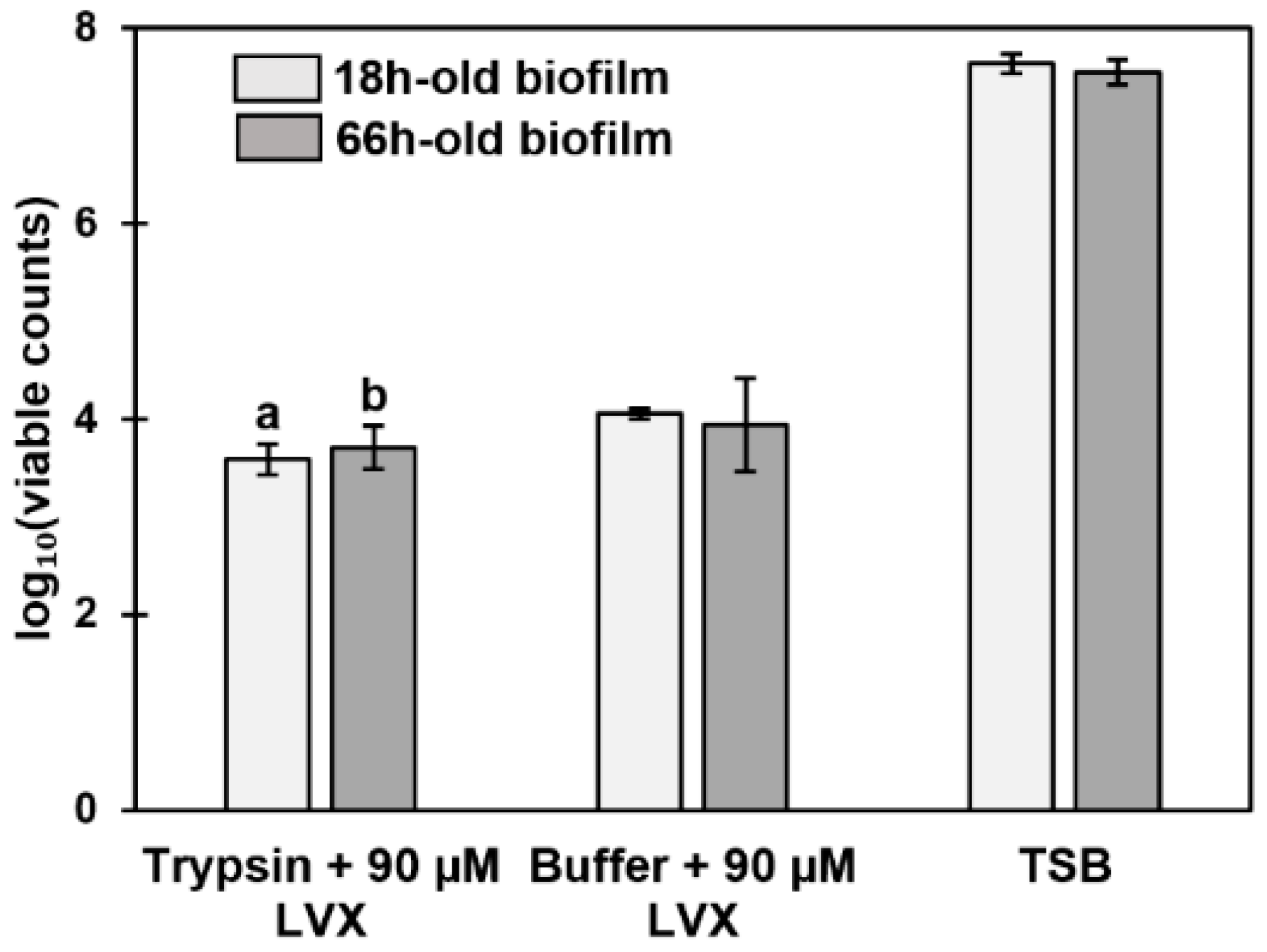
3.7. Antibiotic Susceptibility Depends on the Composition of the Biofilm Surfaceome
4. Discussion
4.1. Structural Features and the Impact of PNAG on Biofilm Growth
4.2. The Accessory and Core Surfaceomes of the S. aureus ATCC 25923 Biofilms
4.3. The Surface-Associated Moonlighters Dominate in All Studied Biofilms
4.4. Older Biofilms Are Not Always More Tolerant Than Younger Biofilms
4.5. Several Biofilm Surfaceome Proteins Are Important for Successful Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nymer, M.; Cope, E.; Brady, R.; Shirtliff, M.E.; Leid, J.G. Immune responses to indwelling medical devices. In The Role of Biofilms in Device-Related Infections, 1st ed.; Shirtliff, M.E., Leid, J.G., Eds.; Springer: Cham, Switzerland, 2008; Volume 3, pp. 239–264. [Google Scholar]
- Zimmerli, W. Clinical presentation and treatment of orthopaedic implant-associated infection. J. Intern. Med. 2014, 276, 111–119. [Google Scholar] [CrossRef]
- Trampuz, A.; Zimmerli, W. Diagnosis and treatment of implant-associated septic arthritis and osteomyelitis. Curr. Infect. Dis. Rep. 2008, 10, 394–403. [Google Scholar] [CrossRef]
- Peters, G.; Locci, R.; Pulverer, G. Adherence and growth of coagulase-negative staphylococci on surfaces of intravenous catheters. J. Infect. Dis. 1982, 146, 479–482. [Google Scholar] [CrossRef]
- Bouza, E.; San Juan, R.; Muñoz, P.; Pascau, J.; Voss, A.; Desco, M.; Cooperative Group of the European Study Group on Nosocomial Infections (ESGNI). A European perspective on intravascular catheter-related infections: Report on the microbiology workload, aetiology and antimicrobial susceptibility (ESGNI-005 Study). Clin. Microbiol. Infect. 2004, 10, 838–842. [Google Scholar] [CrossRef]
- Chua, J.D.; Wilkoff, B.L.; Lee, I.; Juratli, N.; Longworth, D.L.; Gordon, S.M. Diagnosis and management of infections involving implantable electrophysiologic cardiac devices. Ann. Intern. Med. 2000, 133, 604–608. [Google Scholar] [CrossRef]
- Brand, K.G. Infection of mammary prostheses: A survey and the question of prevention. Ann. Plast. Surg. 1993, 30, 289–295. [Google Scholar] [CrossRef]
- Pittet, B.; Montandon, D.; Pittet, D. Infection in breast implants. Lancet Infect. Dis. 2005, 5, 94–106. [Google Scholar] [CrossRef]
- Benito, N.; Franco, M.; Ribera, A.; Soriano, A.; Rodriguez-Pardo, D.; Sorlí, L.; Fresco, G.; Fernández-Sampedro, M.; Dolores Del Toro, M.; Guío, L.; et al. Time trends in the aetiology of prosthetic joint infections: A multicentre cohort study. Clin. Microbiol. Infect. 2016, 22, e1–e8. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef]
- Donlan, R.M. Role of biofilms in antimicrobial resistance. ASAIO J. 2000, 46, S47–S52. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Jensen, P.Ø.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Høiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Ciofu, O.; Molin, S.; Givskov, M.; Høiby, N. Applying insights from biofilm biology to drug development—Can a new approach be developed? Nat. Rev. Drug Discov. 2013, 12, 791–808. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Massey, R.C.; Kantzanou, M.N.; Fowler, T.; Day, N.P.; Schofield, K.; Wann, E.R.; Berendt, A.R.; Höök, M.; Peacock, S.J. Fibronectin-Binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to fibronectin and invasion of endothelial cells. Cell. Microbiol. 2001, 3, 839–851. [Google Scholar] [CrossRef]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Moser, C.; Bassi, G.L.; Coenye, T.; Donelli, G.; Hall-Stoodley, L.; Holá, V.; Imbert, C.; Kirketerp-Møller, K.; et al. ESCMID guideline for the diagnosis and treatment of biofilm infections 2014. Clin. Microbiol. Infect. 2015, 21, 1–25. [Google Scholar] [CrossRef]
- Spellberg, B.; Lipsky, B.A. Systemic antibiotic therapy for chronic osteomyelitis in adults. Clin. Infect. Dis. 2012, 54, 393–407. [Google Scholar] [CrossRef]
- Gergely, I.; Zazgyva, A.; Man, A.; Zuh, S.G.; Pop, T.S. The in vitro antibacterial effect of S53P4 bioactive glass and gentamicin impregnated polymethylmethacrylate beads. Acta Microbiol. Immunol. Hung. 2014, 61, 145–160. [Google Scholar] [CrossRef]
- Drago, L.; Vassena, C.; Fenu, S.; De Vecchi, E.; Signori, V.; De Francesco, R.; Romanò, C.L. In vitro antibiofilm activity of bioactive glass S53P4. Future Microbiol. 2014, 9, 593–601. [Google Scholar] [CrossRef]
- Drago, L.; Boot, W.; Dimas, K.; Malizos, K.; Hänsch, G.M.; Stuyck, J.; Gawlitta, D.; Romanò, C.L. Does implant coating with antibacterial-loaded hydrogel reduce bacterial colonization and biofilm formation in vitro? Clin. Orthop. Relat. Res. 2014, 472, 3311–3323. [Google Scholar] [CrossRef]
- Braem, A.; De Cremer, K.; Delattin, N.; De Brucker, K.; Neirinck, B.; Vandamme, K.; Martens, J.A.; Michiels, J.; Vleugels, J.; Cammue, B.P.; et al. Novel anti-infective implant substrates: Controlled release of antibiofilm compounds from mesoporous silica-containing macroporous titanium. Colloids Surf. B Biointerfaces 2015, 126, 481–488. [Google Scholar] [CrossRef]
- Jennings, J.A.; Carpenter, D.P.; Troxel, K.S.; Beenken, K.E.; Smeltzer, M.S.; Courtney, H.S.; Haggard, W.O. Novel antibiotic-loaded point-of-care implant coating inhibits biofilm. Clin. Orthop. Relat. Res. 2015, 473, 2270–2282. [Google Scholar] [CrossRef]
- Shukla, V.; Bhathena, Z. Sustained release of a purified tannin component of Terminalia chebula from a titanium implant surface prevents biofilm formation by Staphylococcus aureus. Appl. Biochem. Biotechnol. 2015, 175, 3542–3556. [Google Scholar] [CrossRef]
- Tran, N.; Kelley, M.N.; Tran, P.A.; Garcia, D.R.; Jarrell, J.D.; Hayda, R.A.; Born, C.T. Silver doped titanium oxide-PDMS hybrid coating inhibits Staphylococcus aureus and Staphylococcus epidermidis growth on PEEK. Mater. Sci. Eng. C Mater. Biol. Appl. 2015, 49, 201–209. [Google Scholar] [CrossRef]
- Aamdal Scheie, A.; Chamgordani, E.J.; Naemi, A.O.; Hansen, F.K.; Benneche, T. Staphylococcus epidermidis biofilm on implant material is reduced by a covalently linked thiophenone. J. Appl. Microbiol. 2016, 121, 547–553. [Google Scholar] [CrossRef]
- Hiltunen, A.K.; Skogman, M.E.; Rosenqvist, K.; Juvonen, H.; Ihalainen, P.; Peltonen, J.; Juppo, A.; Fallarero, A. Bioactive glass combined with bisphosphonates provides protection against biofilms formed by the periodontal pathogen Aggregatibacter actinomycetemcomitans. Int. J. Pharm. 2016, 501, 211–220. [Google Scholar] [CrossRef]
- Hiltunen, A.K.; Vuorela, P.M.; Fallarero, A. Bisphosphonates offer protection against prosthetic joint infections caused by Staphylococcus aureus and Staphylococcus epidermidis biofilms. J. Drug Deliv. Sci. Technol. 2017, 40, 136–141. [Google Scholar] [CrossRef]
- Liu, X.; Tian, A.; You, J.; Zhang, H.; Wu, L.; Bai, X.; Lei, Z.; Shi, X.; Xue, X.; Wang, H. Antibacterial abilities and biocompatibilities of Ti-Ag alloys with nanotubular coatings. Int. J. Nanomed. 2016, 11, 5743–5755. [Google Scholar] [CrossRef]
- Sankar, G.G.; Murthy, P.S.; Das, A.; Sathya, S.; Nankar, R.; Venugopalan, V.P.; Doble, M. Polydimethyl siloxane based nanocomposites with antibiofilm properties for biomedical applications. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1075–1082. [Google Scholar] [CrossRef]
- Zaatreh, S.; Haffner, D.; Strauß, M.; Wegner, K.; Warkentin, M.; Lurtz, C.; Zamponi, C.; Mittelmeier, W.; Kreikemeyer, B.; Willumeit-Römer, R.; et al. Fast corroding, thin magnesium coating displays antibacterial effects and low cytotoxicity. Biofouling 2017, 33, 294–305. [Google Scholar] [CrossRef]
- Drago, L.; Bortolin, M.; De Vecchi, E.; Agrappi, S.; Weinstein, R.L.; Mattina, R.; Francetti, L. Antibiofilm activity of sandblasted and laser-modified titanium against microorganisms isolated from peri-implantitis lesions. J. Chemother. 2016, 28, 383–389. [Google Scholar] [CrossRef]
- Sánchez, M.C.; Llama-Palacios, A.; Fernández, E.; Figuero, E.; Marín, M.J.; León, R.; Blanc, V.; Herrera, D.; Sanz, M. An in vitro biofilm model associated to dental implants: Structural and quantitative analysis of in vitro biofilm formation on different dental implant surfaces. Dent. Mater. 2014, 30, 1161–1171. [Google Scholar] [CrossRef]
- Pita, P.P.; Rodrigues, J.A.; Ota-Tsuzuki, C.; Miato, T.F.; Zenobio, E.G.; Giro, G.; Figueiredo, L.C.; Gonçalves, C.; Gehrke, S.A.; Cassoni, A.; et al. Oral streptococci biofilm formation on different implant surface topographies. Biomed. Res. Int. 2015, 2015, 159625. [Google Scholar] [CrossRef]
- Al-Ahmad, A.; Karygianni, L.; Schulze Wartenhorst, M.; Bächle, M.; Hellwig, E.; Follo, M.; Vach, K.; Han, J.S. Bacterial adhesion and biofilm formation on yttria-stabilized, tetragonal zirconia and titanium oral implant materials with low surface roughness—An in situ study. J. Med. Microbiol. 2016, 65, 596–604. [Google Scholar] [CrossRef] [Green Version]
- De Avila, E.D.; Avila-Campos, M.J.; Vergani, C.E.; Spolidório, D.M.; Mollo Fde, A., Jr. Structural and quantitative analysis of a mature anaerobic biofilm on different implant abutment surfaces. J. Prosthet. Dent. 2016, 115, 428–436. [Google Scholar] [CrossRef]
- Roehling, S.; Astasov-Frauenhoffer, M.; Hauser-Gerspach, I.; Braissant, O.; Woelfler, H.; Waltimo, T.; Kniha, H.; Gahlert, M. In vitro biofilm formation on titanium and zirconia implant surfaces. J. Periodontol. 2017, 88, 298–307. [Google Scholar] [CrossRef]
- Payne, D.E.; Boles, B.R. Emerging interactions between matrix components during biofilm development. Curr. Genet. 2016, 62, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koseki, H.; Yonekura, A.; Shida, T.; Yoda, I.; Horiuchi, H.; Morinaga, Y.; Yanagihara, K.; Sakoda, H.; Osaki, M.; Tomita, M. Early staphylococcal biofilm formation on solid orthopaedic implant materials: In vitro study. PLoS ONE 2014, 9, e107588. [Google Scholar] [CrossRef]
- Patel, S.S.; Aruni, W.; Inceoglu, S.; Akpolat, Y.T.; Botimer, G.D.; Cheng, W.K.; Danisa, O.A. A comparison of Staphylococcus aureus biofilm formation on cobalt-chrome and titanium-alloy spinal implants. J. Clin. Neurosci. 2016, 31, 219–223. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulston, L.; Elsholz, A.K.; DeFrancesco, A.S.; Losick, R. The extracellular matrix of Staphylococcus aureus biofilms comprises cytoplasmic proteins that associate with the cell surface in response to decreasing pH. MBio 2014, 5, e01667-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speziale, P.; Pietrocola, G.; Foster, T.J.; Geoghegan, J.A. Protein-Based biofilm matrices in staphylococci. Front. Cell. Infect. Microbiol. 2014, 4, 171. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Chang, J.; Rimal, B.; Yang, H.; Schaefer, J. Surface proteins and the formation of biofilms by Staphylococcus aureus. Biochim. Biophys. Acta Biomembr. 2018, 1860, 749–756. [Google Scholar] [CrossRef]
- Porayath, C.; Suresh, M.K.; Biswas, R.; Nair, B.G.; Mishra, N.; Pal, S. Autolysin mediated adherence of Staphylococcus aureus with fibronectin, gelatin and heparin. Int. J. Biol. Macromol. 2018, 110, 179–184. [Google Scholar] [CrossRef]
- Graf, A.C.; Leonard, A.; Schäuble, M.; Rieckmann, L.M.; Hoyer, J.; Maass, S.; Lalk, M.; Becher, D.; Pané-Farré, J.; Riedel, K. Virulence factors produced by Staphylococcus aureus biofilms have a moonlighting function contributing to biofilm integrity. Mol. Cell Proteom. 2019, 18, 1036–1053. [Google Scholar] [CrossRef]
- Lei, M.G.; Gupta, R.K.; Lee, C.Y. Proteomics of Staphylococcus aureus biofilm matrix in a rat model of orthopedic implant-associated infection. PLoS ONE 2017, 12, e0187981. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Maybank, R.A.; Enke, S.; Friss, M.B.; Diviak, L.F.; Karaolis, D.K.; Koren, S.; Ondov, B.; Phillippy, A.M.; Bergman, N.H.; et al. Complete genome sequence of the quality control strain Staphylococcus aureus subsp. aureus ATCC 25923. Genome Announc. 2014, 2, e01110-14. [Google Scholar] [CrossRef] [Green Version]
- Hench, L.L.; Day, D.E.; Höland, W.; Rheinberger, V.M. Glass and medicine. Int. J. Appl. Glass Sci. 2010, 1, 104–117. [Google Scholar] [CrossRef]
- Peters, W.; Fornasier, V. Complications from injectable materials used for breast augmentation. Can. J. Plast. Surg. 2009, 17, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Arora, M.; Chan, E.K.; Gupta, S.; Diwan, A.D. Polymethylmethacrylate bone cements and additives: A review of the literature. World J. Orthop. 2013, 4, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Williams, R.; Aojula, A.; O’Neill, J.; Trzińscka, Z.; Grover, L.; Scott, R.A.; Peacock, A.F.; Logan, A.; Stamboulis, A.; et al. Peptide aptamers: Novel coatings for orthopaedic implants. Mater. Sci. Eng. C Mater. Biol. Appl. 2015, 54, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Skogman, M.E.; Vuorela, P.M.; Fallarero, A. Combining biofilm matrix measurements with biomass and viability assays in susceptibility assessments of antimicrobials against Staphylococcus aureus biofilms. J. Antibiot. 2012, 65, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiltunen, A.; Skogman, M.; Vuorela, P.M.; Fallarero, A. Exploration of microbial communities using the Thermo Scientific Varioskan LUX multimode reader and the invitrogen EVOS FL cell imaging system. Biotechniques 2017, 63, 236–237. [Google Scholar] [CrossRef] [Green Version]
- Savijoki, K.; Nyman, T.A.; Kainulainen, V.; Miettinen, I.; Siljamäki, P.; Fallarero, A.; Sandholm, J.; Satokari, R.; Varmanen, P. Growth mode and carbon source impact the surfaceome dynamics of Lactobacillus rhamnosus GG. Front. Microbiol. 2019, 10, 1272. [Google Scholar] [CrossRef] [Green Version]
- Espino, E.; Koskenniemi, K.; Mato-Rodriguez, L.; Nyman, T.A.; Reunanen, J.; Koponen, J.; Öhman, T.; Siljamäki, P.; Alatossava, T.; Varmanen, P.; et al. Uncovering surface-exposed antigens of Lactobacillus rhamnosus by cell shaving proteomics and two-dimensional immunoblotting. J. Proteome Res. 2015, 14, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Lorey, M.B.; Rossi, K.; Eklund, K.K.; Nyman, T.A.; Matikainen, S. Global characterization of protein secretion from human macrophages following non-canonical caspase-4/5 inflammasome activation. Mol. Cell. Proteom. 2017, 16, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.—Range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Pitts, B.; Hamilton, M.A.; Zelver, N.; Stewart, P.S. A microtiter-plate screening method for biofilm disinfection and removal. J. Microbiol. Methods 2003, 54, 269–276. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bollini, S.; Herbst, J.J.; Gaughan, G.T.; Verdoorn, T.A.; Ditta, J.; Dubowchik, G.M.; Vinitsky, A. High-Throughput fluorescence polarization method for identification of FKBP12 ligands. J. Biomol. Screen. 2002, 7, 526–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, D.; Fischer, W.; Krokotsch, A.; Leopold, K.; Hartmann, R.; Egge, H.; Laufs, R. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: Purification and structural analysis. J. Bacteriol. 1996, 178, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramton, S.E.; Gerke, C.; Schnell, N.F.; Nichols, W.W.; Götz, F. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect. Immun. 1999, 67, 5427–5433. [Google Scholar]
- Stewart, E.J.; Ganesan, M.; Younger, J.G.; Solomon, M.J. Artificial biofilms establish the role of matrix interactions in staphylococcal biofilm assembly and disassembly. Sci. Rep. 2015, 5, 13081. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.H.; Shu, J.C.; Lin, L.P.; Chong, K.Y.; Cheng, Y.W.; Du, J.F.; Liu, S.T. Elucidating the crucial role of poly n-acetylglucosamine from Staphylococcus aureus in cellular adhesion and pathogenesis. PLoS ONE 2015, 10, e0124216. [Google Scholar] [CrossRef] [Green Version]
- Kropec, A.; Maira-Litran, T.; Jefferson, K.K.; Grout, M.; Cramton, S.E.; Götz, F.; Goldmann, D.A.; Pier, G.B. Poly-N-Acetylglucosamine production in Staphylococcus aureus is essential for virulence in murine models of systemic infection. Infect. Immun. 2005, 73, 6868–6876. [Google Scholar] [CrossRef] [Green Version]
- Vuong, C.; Voyich, J.M.; Fischer, E.R.; Braughton, K.R.; Whitney, A.R.; De Leo, F.R.; Otto, M. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell. Microbiol. 2004, 6, 269–275. [Google Scholar] [CrossRef]
- Costa, A.R.; Henriques, M.; Oliveira, R.; Azeredo, J. The role of polysaccharide intercellular adhesin (PIA) in Staphylococcus epidermidis adhesion to host tissues and subsequent antibiotic tolerance. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Sundaramoorthy, R.; Fyfe, P.K.; Hunter, W.N. Structure of Staphylococcus aureus EsxA suggests a contribution to virulence by action as a transport chaperone and/or adaptor protein. J. Mol. Biol. 2008, 383, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Korea, C.G.; Balsamo, G.; Pezzicoli, A.; Merakou, C.; Tavarini, S.; Bagnoli, F.; Serruto, D.; Unnikrishnan, M. Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect. Immun. 2014, 82, 4144–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonesson, A.; Przybyszewska, K.; Eriksson, S.; Mörgelin, M.; Kjellström, S.; Davies, J.; Potempa, J.; Schmidtchen, A. Identification of bacterial biofilm and the Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci. Rep. 2017, 7, 8689. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.R.; Horsburgh, M.J.; Hayhurst, E.J.; Wright, L.; Jonsson, I.M.; Tarkowski, A.; Kokai-Kun, J.F.; Mond, J.J.; Foster, S.J. Characterization of IsaA and SceD, two putative lytic transglycosylases of Staphylococcus aureus. J. Bacteriol. 2007, 189, 7316–7325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.F.; Cheng, J.S.; Sy, C.L.; Huang, W.C.; Yang, H.C.; Gallo, R.L.; Huang, C.M.; Shu, C.W. IsaB Inhibits autophagic flux to promote host transmission of methicillin-resistant Staphylococcus aureus. J. Investig. Dermatol. 2015, 135, 2714–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürgers, R.; Morsczeck, C.; Felthaus, O.; Gosau, M.; Beck, H.C.; Reichert, T.E. Induced surface proteins of Staphylococcus epidermidis adhering to titanium implant substrata. Clin. Oral Investig. 2018, 22, 2663–2668. [Google Scholar] [CrossRef] [Green Version]
- Zoll, S.; Pätzold, B.; Schlag, M.; Götz, F.; Kalbacher, H.; Stehle, T. Structural basis of cell wall cleavage by a staphylococcal autolysin. PLoS Pathog. 2010, 6, e1000807. [Google Scholar] [CrossRef] [Green Version]
- Houston, P.; Rowe, S.E.; Pozzi, C.; Waters, E.M.; O’Gara, J.P. Essential Role for the Major Autolysin in the Fibronectin-Binding Protein-Mediated Staphylococcus aureus Biofilm Phenotype. Infect. Immun. 2011, 79, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Livesley, M.A.; Lambert, P.A.; Littler, W.A.; Elliott, T.S. Identification of a novel antigen from Staphylococcus epidermidis. FEMS Immunol. Med. Microbiol. 2000, 29, 213–220. [Google Scholar] [CrossRef]
- Resch, A.; Rosenstein, R.; Nerz, C.; Götz, F. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl. Environ. Microbiol. 2005, 71, 2663–2676. [Google Scholar] [CrossRef] [Green Version]
- Merino, N.; Toledo-Arana, A.; Vergara-Irigaray, M.; Valle, J.; Solano, C.; Calvo, E.; Lopez, J.A.; Foster, T.J.; Penadés, J.R.; Lasa, I. Protein A-mediated multicellular behavior in Staphylococcus aureus. J. Bacteriol. 2009, 191, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zabad, S.; Liu, H.; Wang, W.; Jeffery, C. MoonProt 2.0: An expansion and update of the moonlighting proteins database. Nucleic Acids Res. 2018, 46, D640–D644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Serrano, L.; Hernández, S.; Calvo, A.; Severi, M.A.; Ferragut, G.; Pérez-Pons, J.A.; Piñol, J.; Pich, Ò.; Mozo-Villarias, Á.; Amela, I.; et al. MultitaskProtDB-II: An update of a database of multitasking/moonlighting proteins. Nucleic Acids Res. 2018, 46, D645–D648. [Google Scholar] [CrossRef] [PubMed]
- Ebner, P.; Rinker, J.; Götz, F. Excretion of cytoplasmic proteins in Staphylococcus is most likely not due to cell lysis. Curr. Genet. 2016, 62, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.C.; Mann, E.E.; Endres, J.L.; Weiss, E.C.; Cassat, J.E.; Smeltzer, M.S.; Bayles, K.W. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8113–8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashruwala, A.A.; Guchte, A.V.; Boyd, J.M. Impaired respiration elicits SrrAB-dependent programmed cell lysis and biofilm formation in Staphylococcus aureus. eLife 2017, 6, e23845. [Google Scholar] [CrossRef] [PubMed]
- Pasztor, L.; Ziebandt, A.K.; Nega, M.; Schlag, M.; Haase, S.; Franz-Wachtel, M.; Madlung, J.; Nordheim, A.; Heinrichs, D.E.; Götz, F. Staphylococcal major autolysin (Atl) is involved in excretion of cytoplasmic proteins. J. Biol. Chem. 2010, 285, 36794–36803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.K.; Zhang, X.Z.; Lu, C.D.; Tai, P.C. An internal hydrophobic helical domain of Bacillus subtilis enolase is essential but not sufficient as a non-cleavable signal for its secretion. Biochem. Biophys. Res. Commun. 2014, 446, 901–905. [Google Scholar] [CrossRef] [Green Version]
- Widjaja, M.; Harvey, K.L.; Hagemann, L.; Berry, I.J.; Jarocki, V.M.; Raymond, B.B.A.; Tacchi, J.L.; Gründel, A.; Steele, J.R.; Padula, M.P.; et al. Elongation factor tu is a multifunctional and processed moonlighting protein. Sci. Rep. 2017, 7, 11227. [Google Scholar] [CrossRef] [Green Version]
- Kainulainen, V.; Korhonen, T.K. Dancing to another tune-adhesive moonlighting proteins in bacteria. Biology 2014, 3, 178–204. [Google Scholar] [CrossRef] [Green Version]
- Rieu, A.; Aoudia, N.; Jego, G.; Chluba, J.; Yousfi, N.; Briandet, R.; Deschamps, J.; Gasquet, B.; Monedero, V.; Garrido, C.; et al. The biofilm mode of life boosts the anti-inflammatory properties of Lactobacillus. Cell. Microbiol. 2014, 16, 1836–1853. [Google Scholar] [CrossRef]
- Arita-Morioka, K.; Yamanaka, K.; Mizunoe, Y.; Ogura, T.; Sugimoto, S. Novel strategy for biofilm inhibition by using small molecules targeting molecular chaperone DnaK. Antimicrob. Agents Chemother. 2015, 59, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vastano, V.; Pagano, A.; Fusco, A.; Merola, G.; Sacco, M.; Donnarumma, G. The Lactobacillus plantarum EnoA1 enolase is involved in immunostimulation of caco-2 cells and in biofilm development. Adv. Exp. Med. Biol. 2016, 897, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Daubenspeck, J.M.; Liu, R.; Dybvig, K. Rhamnose links moonlighting proteins to membrane phospholipid in Mycoplasmas. PLoS ONE 2016, 11, e0162505. [Google Scholar] [CrossRef] [PubMed]
- Alreshidi, M.M.; Dunstan, R.H.; Gottfries, J.; Macdonald, M.M.; Crompton, M.J.; Ang, C.S.; Williamson, N.A.; Roberts, T.K. Changes in the cytoplasmic composition of amino acids and proteins observed in Staphylococcus aureus during growth under variable growth conditions representative of the human wound site. PLoS ONE 2016, 11, e0159662. [Google Scholar] [CrossRef] [Green Version]
- Voigt, B.; Albrecht, D.; Dalhoff, A. Mode of action of MCB3681 in Staphylococcus aureus—A proteomic study. Arch. Clin. Microbiol. 2016, 7, 31. [Google Scholar] [CrossRef]
- Richardson, A.R.; Somerville, G.A.; Sonenshein, A.L. Regulating the intersection of metabolism and pathogenesis in gram-positive bacteria. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P.; et al. Gram-Positive bacteria produce membrane vesicles: Proteomics-Based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef]
- Lee, J.; Lee, E.Y.; Kim, S.H.; Kim, D.K.; Park, K.S.; Kim, K.P.; Kim, Y.K.; Roh, T.Y.; Gho, Y.S. Staphylococcus aureus extracellular vesicles carry biologically active β-lactamase. Antimicrob. Agents Chemother. 2013, 57, 2589–2595. [Google Scholar] [CrossRef] [Green Version]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [Green Version]
- Gurung, M.; Moon, D.C.; Choi, C.W.; Lee, J.H.; Bae, Y.C.; Kim, J.; Lee, Y.C.; Seol, S.Y.; Cho, D.T.; Kim, S.I.; et al. Staphylococcus aureus produces membrane-derived vesicles that induce host cell death. PLoS ONE 2011, 6, e27958. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.; Oh, M.H.; Jun, S.H.; Kim, S.I.; Choi, C.W.; Kwon, H.I.; Na, S.H.; Kim, Y.J.; Nicholas, A.; Selasi, G.N.; et al. Variation among Staphylococcus aureus membrane vesicle proteomes affects cytotoxicity of host cells. Microb. Pathog. 2016, 93, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Maidhof, H.; Reinicke, B.; Blümel, P.; Berger-Bächi, B.; Labischinski, H. FemA, which encodes a factor essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains. J. Bacteriol. 1991, 173, 3507–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henze, U.; Sidow, T.; Wecke, J.; Labischinski, H.; Berger-Bächi, B. Influence of femB on methicillin resistance and peptidoglycan metabolism in Staphylococcus aureus. J Bacteriol. 1993, 175, 1612–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Hunter, H.N.; Prova, S.; Verma, V.; Qamar, A.; Golemi-Kotra, D. The Staphylococcus aureus methicillin resistance factor FmtA is a D-amino esterase that acts on teichoic acids. mBio 2016, 7, e02070-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courvalin, P. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 2006, 42, S25–S34. [Google Scholar] [CrossRef]
- Mirelman, D.; Sharon, N. Biosynthesis of peptidoglycan by a cell wall preparation of Staphylococcus aureus and its inhibition by penicillin. Biochem. Biophys. Res. Commun. 1972, 46, 1909–1917. [Google Scholar] [CrossRef]
- Valentín, S.; Morales, A.; Sánchez, J.L.; Rivera, A. Safety and efficacy of doxycycline in the treatment of rosacea. Clin. Cosmet. Investig. Dermatol. 2009, 2, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Hooper, D.C. Mode of action of fluoroquinolones. Drugs 1999, 58, 6–10. [Google Scholar] [CrossRef]
- Li, C.; Renz, N.; Trampuz, A. Management of periprosthetic joint infection. Hip Pelvis 2018, 30, 138–146. [Google Scholar] [CrossRef]
- Manner, S.; Goeres, D.M.; Skogman, M.; Vuorela, P.; Fallarero, A. Prevention of Staphylococcus aureus biofilm formation by antibiotics in 96-microtiter well plates and drip flow reactors: Critical factors influencing outcomes. Sci. Rep. 2017, 7, 43854. [Google Scholar] [CrossRef] [Green Version]
- Mandell, J.B.; Orr, S.; Koch, J.; Nourie, B.; Ma, D.; Bonar, D.D.; Shah, N.; Urish, K.L. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J. Orthop. Res. 2019, 37, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Michalik, S.; Depke, M.; Murr, A.; Gesell Salazar, M.; Kusebauch, U.; Sun, Z.; Meyer, T.C.; Surmann, K.; Pförtner, H.; Hildebrandt, P.; et al. A global Staphylococcus aureus proteome resource applied to the in vivo characterization of host-pathogen interactions. Sci. Rep. 2017, 7, 9718. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Chastanet, A.; Qazi, S.; Sørensen, K.; Hill, P.; Msadek, T.; Ingmer, H. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol. Microbiol. 2004, 54, 1445–1462. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Sdr (%) | Vv (µm3/µm2) |
|---|---|---|
| Borosilicate glass | 0.3 ± 0.1 | 0.0048 ± 0.001 |
| Plexiglass | 123 ± 20 | 0.62 ± 0.07 |
| Hydroxyapatite | 58 ± 10 | 0.75 ± 0.08 |
| Titanium | 9.0 ± 1.1 | 0.19 ± 0.02 |
| Polystyrene | 3.0 ± 0.4 | 0.012 ± 0.002 |
| Protein Name | Acc. No. a | 18 h | 42 h | 66 h | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PS | G | HA | PG | TI | PS | G | HA | PG | TI | PS | G | HA | PG | TI | ||
| Bifunctional autolysin—AtlA | Q6GI31 | |||||||||||||||
| Immunoglobulin G-binding protein A—Spa | P38507 | |||||||||||||||
| Immunoglobulin-binding protein—Sbi | Q6GE15 | |||||||||||||||
| Immunodominant antigen B—IsaB | Q6GDG4 | |||||||||||||||
| Immunodominant antigen A—IsaA | Q6GDN1 | |||||||||||||||
| Malate:quinone oxidoreductase 2—MQO2 | Q6GDJ6 | |||||||||||||||
| Leukocidin-like protein 2—Luk2 | Q6GF49 | |||||||||||||||
| Leukocidin-like protein 1—Luk1 | Q6GF50 | |||||||||||||||
| Non-heme ferritin—FtnA | Q99SZ3 | |||||||||||||||
| Foldase protein A—PrsA | Q6GFL5 | |||||||||||||||
| Thioredoxin | Q6GHU0 | |||||||||||||||
| Staphylococcal secretory antigen—SsaA | Q99RX4 | |||||||||||||||
| Putative dipeptidase SAR1836 | Q6GFV0 | |||||||||||||||
| Clp protease proteolytic subunit—ClpP | Q6GIM3 | |||||||||||||||
| Clp ATPase ClpC | Q99W78 | |||||||||||||||
| Clp ATPase ClpL | Q6GDQ0 | |||||||||||||||
| Clp ATPase ClpB | Q6GIB2 | |||||||||||||||
| Gamma-hemolysin component B—HlgB | Q6GE12 | |||||||||||||||
| Protein RecA | Q6GHF0 | |||||||||||||||
| Bone sialoprotein-binding protein—Bbp | Q6GJA6 | |||||||||||||||
| Clumping factor B—ClfB | Q6GDH2 | |||||||||||||||
| Clumping factor A—ClfA | Q6GIK4 | |||||||||||||||
| Delta-hemolysin—HglD | Q6GF37 | |||||||||||||||
| Virulence factor—EsxA | Q99WU4 | |||||||||||||||
| ATP-dependent protease ATPase—HslU | Q6GHI1 | |||||||||||||||
| Elastin-binding protein—EbpS | Q6GGT1 | |||||||||||||||
| Fibrinogen-binding protein—FbnBP | Q6GHS9 | |||||||||||||||
| Fibronectin-binding protein A—FnBPA | Q6GDU5 | |||||||||||||||
| Ser-Asp repeat-containing protein C—SdrC | Q6GJA7 | |||||||||||||||
| Ser-Asp repeat-containing protein D—SdrD | Q8NXX6 | |||||||||||||||
| Catabolite control protein A—CcpA | Q6GFX2 | |||||||||||||||
| Response regulator—CodY | Q6GHI0 | |||||||||||||||
| Response regulator—SarA | Q7A732 | |||||||||||||||
| Response regulator—Rot | Q9RFJ6 | |||||||||||||||
| Response regulator—SarR | Q9F0R1 | |||||||||||||||
| Response regulator—SarS | Q7A872 | |||||||||||||||
| Response regulator—VraR | Q7A4R9 | |||||||||||||||
| Response regulator—SaeR | Q99VR7 | |||||||||||||||
| Response regulator—MsrR | Q99Q02 | |||||||||||||||
| Response regulator—MraZ | Q6GHQ7 | |||||||||||||||
| Response regulator—LytR | P52078 | |||||||||||||||
| Response regulator—NrdR | Q6GG20 | |||||||||||||||
| Response regulator—GraR | Q6GJ11 | |||||||||||||||
| HTH-type transcriptional regulator—MgrA | Q99VT5 | |||||||||||||||
| Redox-sensing repressor—Rex | Q6GF26 | |||||||||||||||
| SOS response repressor—LexA | Q9L4P1 | |||||||||||||||
| Oxygen regulatory protein—NreC | Q99RN8 | |||||||||||||||
| Regulatory protein—Spx | Q6GI88 | |||||||||||||||
| Histidine protein kinase—SaeS | Q99VR8 | |||||||||||||||
| RNA polymerase sigma factor SigA | Q99TT5 | |||||||||||||||
| Anti-sigma-B factor antagonist—RsbV | Q6GF07 | |||||||||||||||
| Iron-regulated surface determinant—IsdB | Q6GHV7 | |||||||||||||||
| Lysostaphin resistance protein A—LyrA | Q6GEA0 | |||||||||||||||
| Methicillin-resistance protein—FmtA | Q6GI27 | |||||||||||||||
| Conserved virulence factor B—CvfB | Q99U93 | |||||||||||||||
| DegV domain-containing protein SAR1438 | Q6GGY2 | |||||||||||||||
| Signal transduction protein TRAP | Q6GFM2 | |||||||||||||||
| Staphopain A (cysteine protease)—SspP | Q6GFE8 | |||||||||||||||
| Ferrochelatase—HemH | Q6G8A3 | |||||||||||||||
| Phospholipase C—PlC | Q5HEI1 | |||||||||||||||
| Methicillin resistance-associated—FemA | Q99UA7 | |||||||||||||||
| Methicillin resistance-associated—FemB | Q6GH30 | |||||||||||||||
| Probable cell wall amidase—LytH | Q7A588 | |||||||||||||||
| ATP-dependent protease subunit—HslV | Q6GHI2 | |||||||||||||||
| CtpA-like serine protease | Q6GGY8 | |||||||||||||||
| HtrA-like serine protease | Q6GI62 | |||||||||||||||
| Hydrolase encoded by the agr operon | P55177 | |||||||||||||||
| Probable thiol peroxidase | Q6GFZ4 | |||||||||||||||
| Uncharacterized oxidoreductase SAR2567 | Q6GDV6 | |||||||||||||||
| Peptide methionine sulfoxide reductase MsrB | Q6GGY4 | |||||||||||||||
| Heme-dependent peroxidase (SAV0587) | Q99W24 | |||||||||||||||
| Thioredoxin reductase | Q6GB66 | |||||||||||||||
| NADPH-dependent oxidoreductase | Q6GJR6 | |||||||||||||||
| Multicopper oxidase—Mco | Q6GIX3 | |||||||||||||||
| Nitric oxide synthase oxygenase | Q6GFE2 | |||||||||||||||
| Putative NAD(P)H nitroreductase (SAV2523) | Q99RB2 | |||||||||||||||
| FMN-dependent NADPH-azoreductase | Q99W49 | |||||||||||||||
| Staphylocoagulase—Coa | P17855 | |||||||||||||||
| Iron-sulfur cluster repair protein—ScdA | Q6GK53 | |||||||||||||||
| Urease accessory protein G—UreG | Q99RX9 | |||||||||||||||
| ATP synthase epsilon chain | Q6GEX3 | |||||||||||||||
| ATP synthase subunit delta | Q6GEW9 | |||||||||||||||
| ATP synthase gamma chain | Q99SF4 | |||||||||||||||
| 30S ribosomal protein S1 | Q6GGT5 | |||||||||||||||
| 30S ribosomal protein S10 | Q931G5 | |||||||||||||||
| 30S ribosomal protein S11 | Q6GEK8 | |||||||||||||||
| 30S ribosomal protein S12 | Q6GJC3 | |||||||||||||||
| 30S ribosomal protein S13 | Q6GEK7 | |||||||||||||||
| 30S ribosomal protein S15 | Q99UJ9 | |||||||||||||||
| 30S ribosomal protein S16 | Q6GHJ7 | |||||||||||||||
| 30S ribosomal protein S17 | Q8NVB4 | |||||||||||||||
| 30S ribosomal protein S18 | Q6GJV1 | |||||||||||||||
| 30S ribosomal protein S19 | Q6GEI7 | |||||||||||||||
| 30S ribosomal protein S2 | Q6GHH9 | |||||||||||||||
| 30S ribosomal protein S20 | Q99TR3 | |||||||||||||||
| 30S ribosomal protein S21 | Q6GGC5 | |||||||||||||||
| 30S ribosomal protein S3 | Q6GEI9 | |||||||||||||||
| 30S ribosomal protein S4 | Q6GFY8 | |||||||||||||||
| 30S ribosomal protein S5 | Q6GEK0 | |||||||||||||||
| 30S ribosomal protein S6 | Q6GJV3 | |||||||||||||||
| 30S ribosomal protein S7 | Q6GJC2 | |||||||||||||||
| 30S ribosomal protein S8 | Q6GEJ7 | |||||||||||||||
| 30S ribosomal protein S9 | Q6GEL8 | |||||||||||||||
| 50S ribosomal protein L1 | Q6GJD0 | |||||||||||||||
| 50S ribosomal protein L10 | Q6GJC9 | |||||||||||||||
| 50S ribosomal protein L11 | Q6GJD1 | |||||||||||||||
| 50S ribosomal protein L13 | Q99S51 | |||||||||||||||
| 50S ribosomal protein L14 | Q99S31 | |||||||||||||||
| 50S ribosomal protein L15 | Q6GEK2 | |||||||||||||||
| 50S ribosomal protein L16 | Q99S28 | |||||||||||||||
| 50S ribosomal protein L17 | Q99S46 | |||||||||||||||
| 50S ribosomal protein L18 | Q99S37 | |||||||||||||||
| 50S ribosomal protein L2 | Q6GEI6 | |||||||||||||||
| 50S ribosomal protein L20 | Q6GG27 | |||||||||||||||
| 50S ribosomal protein L21 | Q99TK6 | |||||||||||||||
| 50S ribosomal protein L22 | Q99S26 | |||||||||||||||
| 50S ribosomal protein L23 | Q99S23 | |||||||||||||||
| 50S ribosomal protein L24 | Q6GEJ4 | |||||||||||||||
| 50S ribosomal protein L25 | Q99WA2 | |||||||||||||||
| 50S ribosomal protein L27 | Q931Q3 | |||||||||||||||
| 50S ribosomal protein L28 | Q6GHL1 | |||||||||||||||
| 50S ribosomal protein L29 | Q6GEJ1 | |||||||||||||||
| 50S ribosomal protein L3 | Q6GEI3 | |||||||||||||||
| 50S ribosomal protein L30 | Q6GEK1 | |||||||||||||||
| 50S ribosomal protein L31 | Q6GEV5 | |||||||||||||||
| 50S ribosomal protein L35 | Q6GG26 | |||||||||||||||
| 50S ribosomal protein L4 | Q6GEI4 | |||||||||||||||
| 50S ribosomal protein L5 | Q99S33 | |||||||||||||||
| 50S ribosomal protein L6 | Q99S36 | |||||||||||||||
| 50S ribosomal protein L7/L12 | Q6GJC8 | |||||||||||||||
| 50S ribosomal protein L9 | Q6GKT0 | |||||||||||||||
| Elongation factor Tu—EfTU | Q6GJC0 | |||||||||||||||
| Elongation factor G—EfG | Q6GJC1 | |||||||||||||||
| Translation initiation factor IF-3—InfC | Q6GG25 | |||||||||||||||
| Translation initiation factor IF-2—InfB | Q6GHG6 | |||||||||||||||
| Elongation factor P—EfP | Q6GGH0 | |||||||||||||||
| Glyceraldehyde-3-phosphate dehydrogenase | Q6GIL8 | |||||||||||||||
| Enolase—ENO | Q6GIL4 | |||||||||||||||
| Phosphoglycerate kinase—PGK | Q6GIL7 | |||||||||||||||
| Pyruvate kinase—PYK | Q6GG09 | |||||||||||||||
| Fructose-bisphosphate aldolase class 1—FBA | Q6GDJ7 | |||||||||||||||
| Pyruvate dehydrogenase E1—PDHB | Q6GHZ1 | |||||||||||||||
| Triosephosphate isomerase—TPI | Q6GIL6 | |||||||||||||||
| ATP-dependent 6-phosphofructokinase—PFK | Q6GG08 | |||||||||||||||
| 2,3-phosphoglycerate mutase—PPGM | Q6GE17 | |||||||||||||||
| Aconitase A—AcnA | Q6GH55 | |||||||||||||||
| L-lactate dehydrogenase 1—L-LDH | Q6GK73 | |||||||||||||||
| D-lactate dehydrogenase—D-LDH | Q6GDS2 | |||||||||||||||
| Alkaline shock protein 23—Asp23 | Q6GEP7 | |||||||||||||||
| Alcohol dehydrogenase—ADH | Q99W07 | |||||||||||||||
| Trigger factor—Tf | Q6GG30 | |||||||||||||||
| DNA-directed RNA polymerase—RpoB | Q6GJC5 | |||||||||||||||
| Alkyl hydroperoxide reductase—AhpC | Q6GJR7 | |||||||||||||||
| Alkyl hydroperoxide reductase—AhpF | Q6GJR8 | |||||||||||||||
| Chaperone protein—GroEL | Q6GF43 | |||||||||||||||
| Chaperone protein—DnaK | Q6GGC0 | |||||||||||||||
| Chaperone protein—DnaJ | Q6GGC1 | |||||||||||||||
| 10 kDa chaperonin | Q6GF42 | |||||||||||||||
| Universal stress protein (SAV1710)—Usp | Q99TF3 | |||||||||||||||
| Superoxide dismutase [Mn/Fe] 1—SodA | Q6GGE6 | |||||||||||||||
| DNA mismatch repair protein—MutL | Q93T05 | |||||||||||||||
| Thermonuclease | Q5HHM4 | |||||||||||||||
| Glutamine synthetase | Q6GHC6 | |||||||||||||||
 a Acc. No., accession numbers were retrieved from the UniProt protein database. In red are proteins involved in adherence and/or adherent growth (biofilm formation), and in blue are adhesive moonlighters.
a Acc. No., accession numbers were retrieved from the UniProt protein database. In red are proteins involved in adherence and/or adherent growth (biofilm formation), and in blue are adhesive moonlighters.| Biofilm Age | Biofilm Formed on | Exposure Time | Penicillin G (2.0 µM) a | Levofloxacin (90.0 µM) a | Doxycycline (4.0 μM) a | Vancomycin (5.0 µM) a |
|---|---|---|---|---|---|---|
| 18 h | HA | 2 h | 0.10 ± 0.28 | 0.56 ± 0.06 | 0.08 ± 0.15 | 0.03 ± 0.31 |
| 18 h | TI | 2 h | 0.01 ± 0.19 | 1.39 ± 0.05 ***, ⱡ ⱡ ⱡ | 0.59 ± 0.21 *, ⱡ | −0.17 ± 0.25 |
| 18 h | HA | 24 h | 2.32 ± 0.18 ***, ⱡ ⱡ ⱡ, Ω | 3.09 ± 0.04 ***, ⱡ ⱡ ⱡ, Ω Ω Ω | 2.00 ± 0.14 *, ⱡ, Ω Ω Ω | 0.65 ± 0.09 Ω |
| 18 h | TI | 24 h | 1.28 ± 0.08 ⱡ | 2.18 ± 0.15 ⱡ ⱡ ⱡ, Ω Ω Ω | 1.08 ± 0.20 Ω | 0.33 ± 0.09 |
| 66 h | HA | 2 h | 0.42 ± 0.04 * | 0.64 ± 0.24 *, ⱡ | 0.31 ± 0.08 | 0.61 ± 0.11 *, ⱡ |
| 66 h | TI | 2 h | 0.07 ± 0.15 | 0.46 ± 0.08 | 0.39 ± 0.06 | 0.24 ± 0.12 |
| 66 h | HA | 24 h | 1.82 ± 0.24 *, Ω Ω Ω | 2.35 ± 0.07 ***, Ω | 1.32 ± 0.20 Ω | 0.88 ± 0.38 |
| 66 h | TI | 24 h | 0.94 ± 0.18 Ω | 1.76 ± 0.14 Ω Ω Ω | 1.62 ± 0.04 *, ⱡ, Ω Ω Ω | 1.67 ± 0.03 Ω |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiltunen, A.K.; Savijoki, K.; Nyman, T.A.; Miettinen, I.; Ihalainen, P.; Peltonen, J.; Fallarero, A. Structural and Functional Dynamics of Staphylococcus aureus Biofilms and Biofilm Matrix Proteins on Different Clinical Materials. Microorganisms 2019, 7, 584. https://doi.org/10.3390/microorganisms7120584
Hiltunen AK, Savijoki K, Nyman TA, Miettinen I, Ihalainen P, Peltonen J, Fallarero A. Structural and Functional Dynamics of Staphylococcus aureus Biofilms and Biofilm Matrix Proteins on Different Clinical Materials. Microorganisms. 2019; 7(12):584. https://doi.org/10.3390/microorganisms7120584
Chicago/Turabian StyleHiltunen, Anna K., Kirsi Savijoki, Tuula A. Nyman, Ilkka Miettinen, Petri Ihalainen, Jouko Peltonen, and Adyary Fallarero. 2019. "Structural and Functional Dynamics of Staphylococcus aureus Biofilms and Biofilm Matrix Proteins on Different Clinical Materials" Microorganisms 7, no. 12: 584. https://doi.org/10.3390/microorganisms7120584
APA StyleHiltunen, A. K., Savijoki, K., Nyman, T. A., Miettinen, I., Ihalainen, P., Peltonen, J., & Fallarero, A. (2019). Structural and Functional Dynamics of Staphylococcus aureus Biofilms and Biofilm Matrix Proteins on Different Clinical Materials. Microorganisms, 7(12), 584. https://doi.org/10.3390/microorganisms7120584