Antibiotic-Induced Perturbations Are Manifested in the Dominant Intestinal Bacterial Phyla of Atlantic Salmon



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Fish, Rearing Conditions and Antibiotic Dosing
2.3. Sampling Strategy
2.4. Bacterial DNA Isolation, PCR Amplification, 16S rRNA Gene Amplicon Library Preparation and Sequencing
2.5. Bioinformatic Analysis of the 16S rRNA Gene Sequence Data
2.5.1. Sequence Data Quality Check
2.5.2. Sequence Data Processing
2.5.3. Accession Number
2.5.4. Sequence Data Analysis to Understand the Gut Microbial Diversity and Composition
2.5.5. Statistical Analyses of the Sequence Data
2.6. Microbial Association Graph Construction and Network Topology Inference
3. Results
3.1. Sequence Data and Analyses Strategy
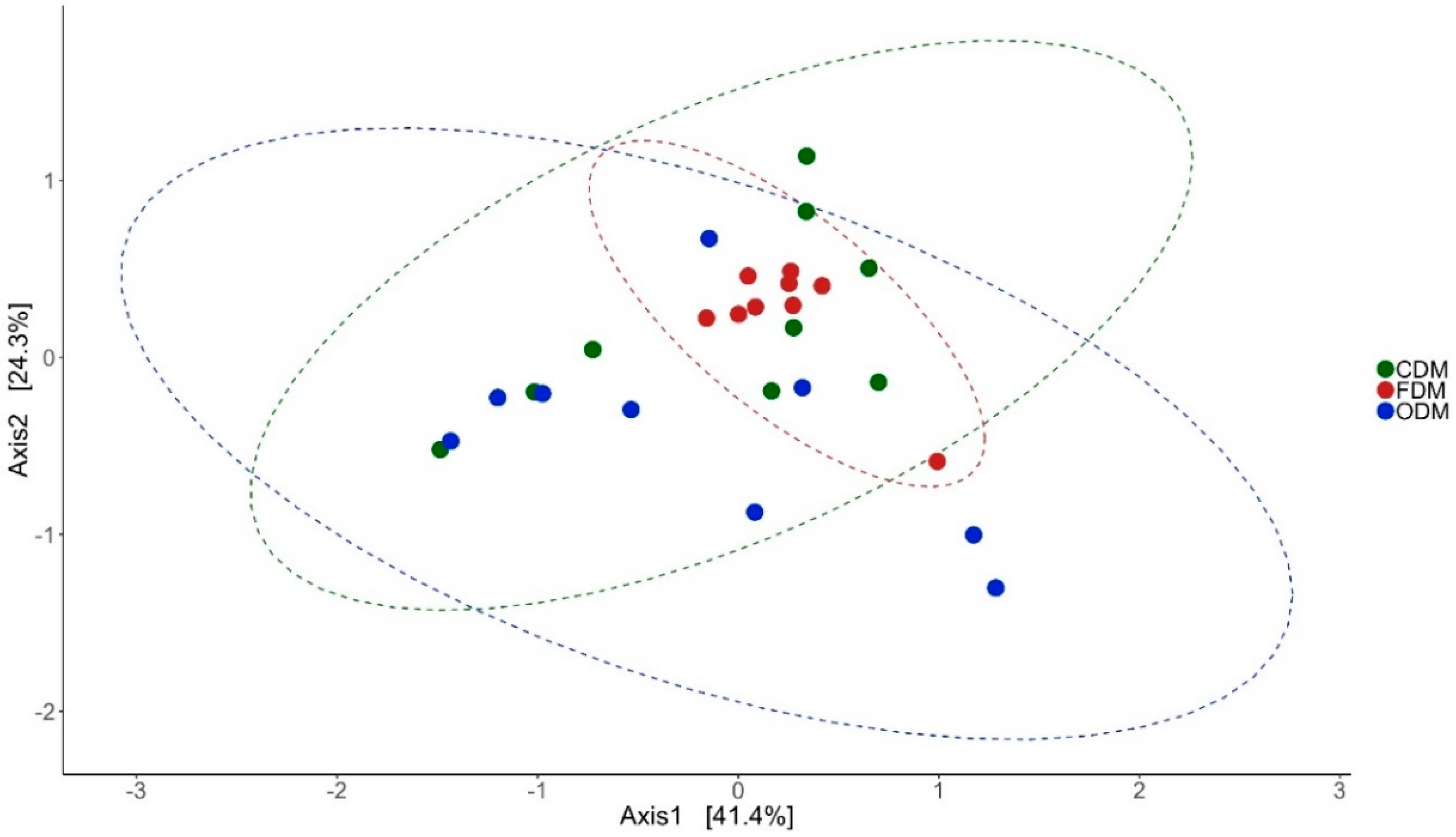
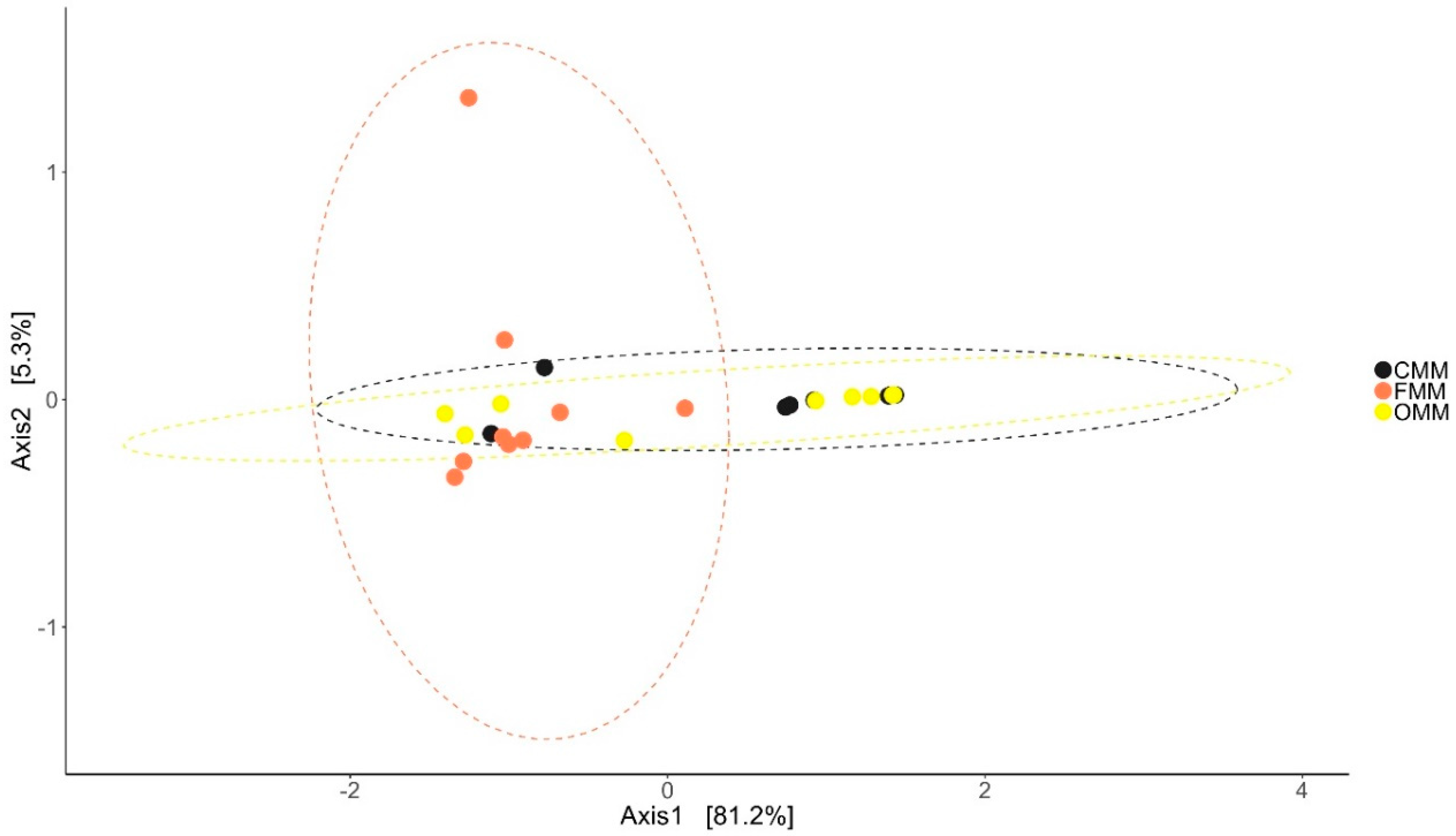
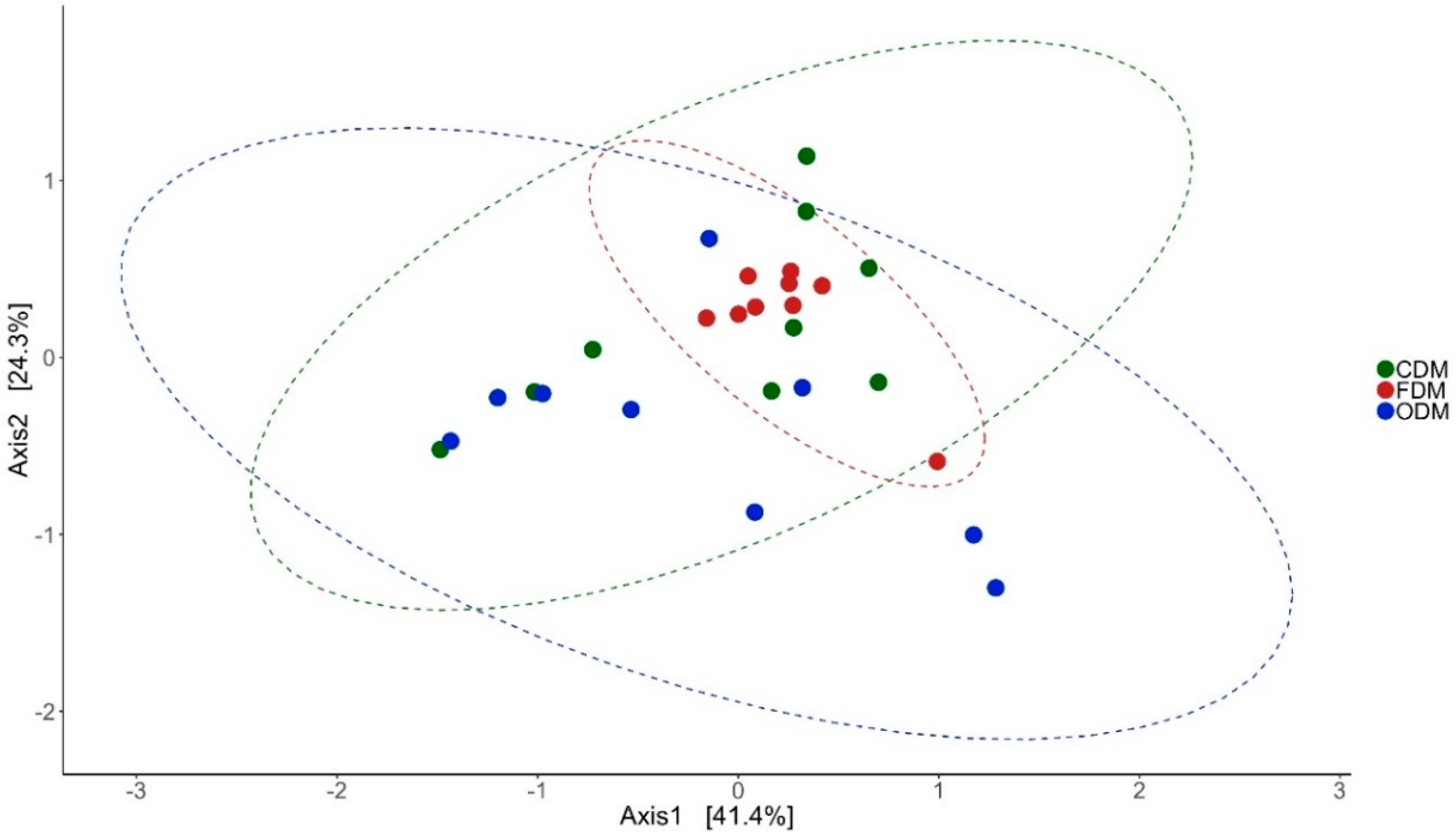
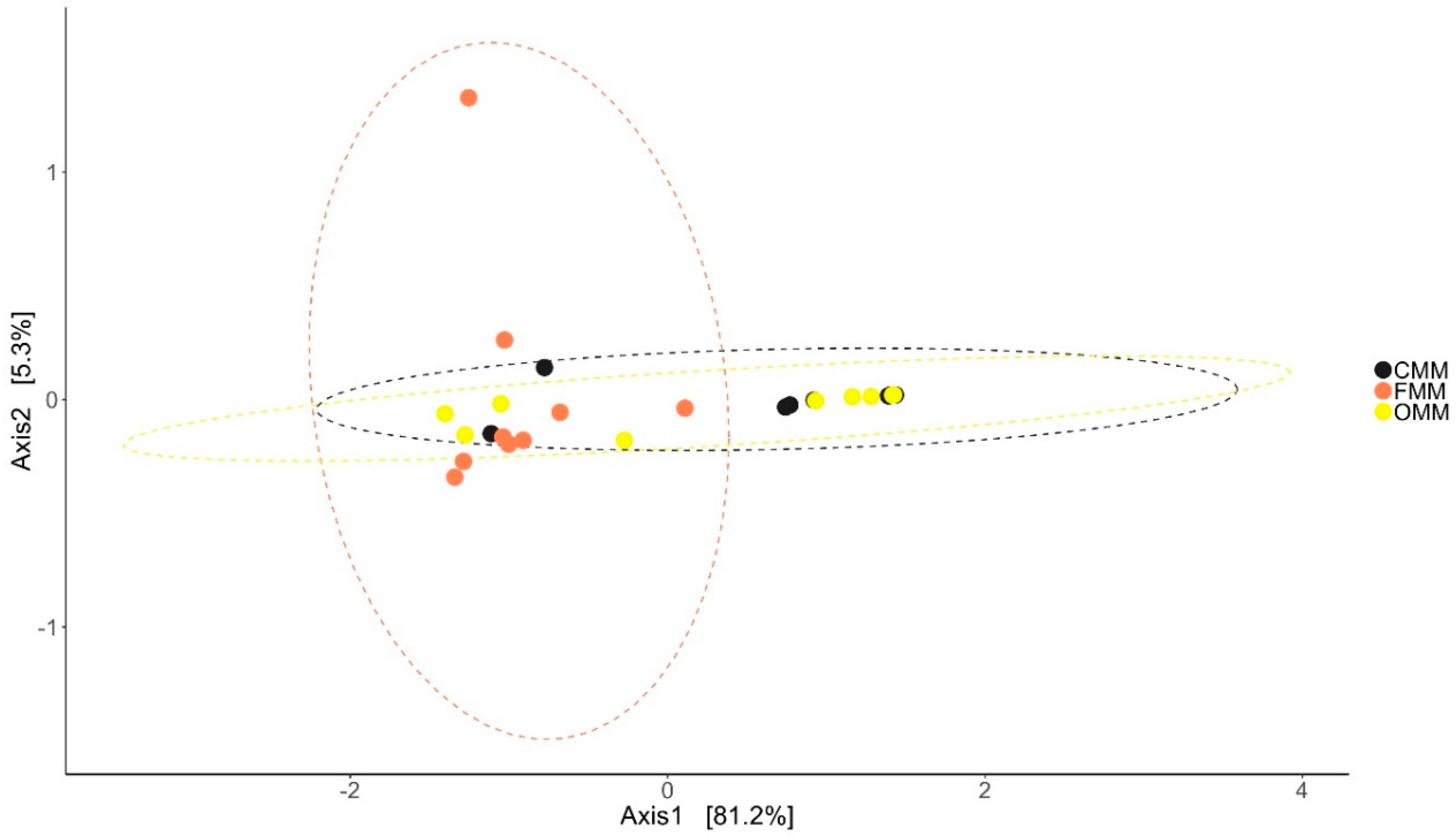
3.2. Changes in the Microbial Diversity of the Intestinal Mucus and Environmental Microbiota
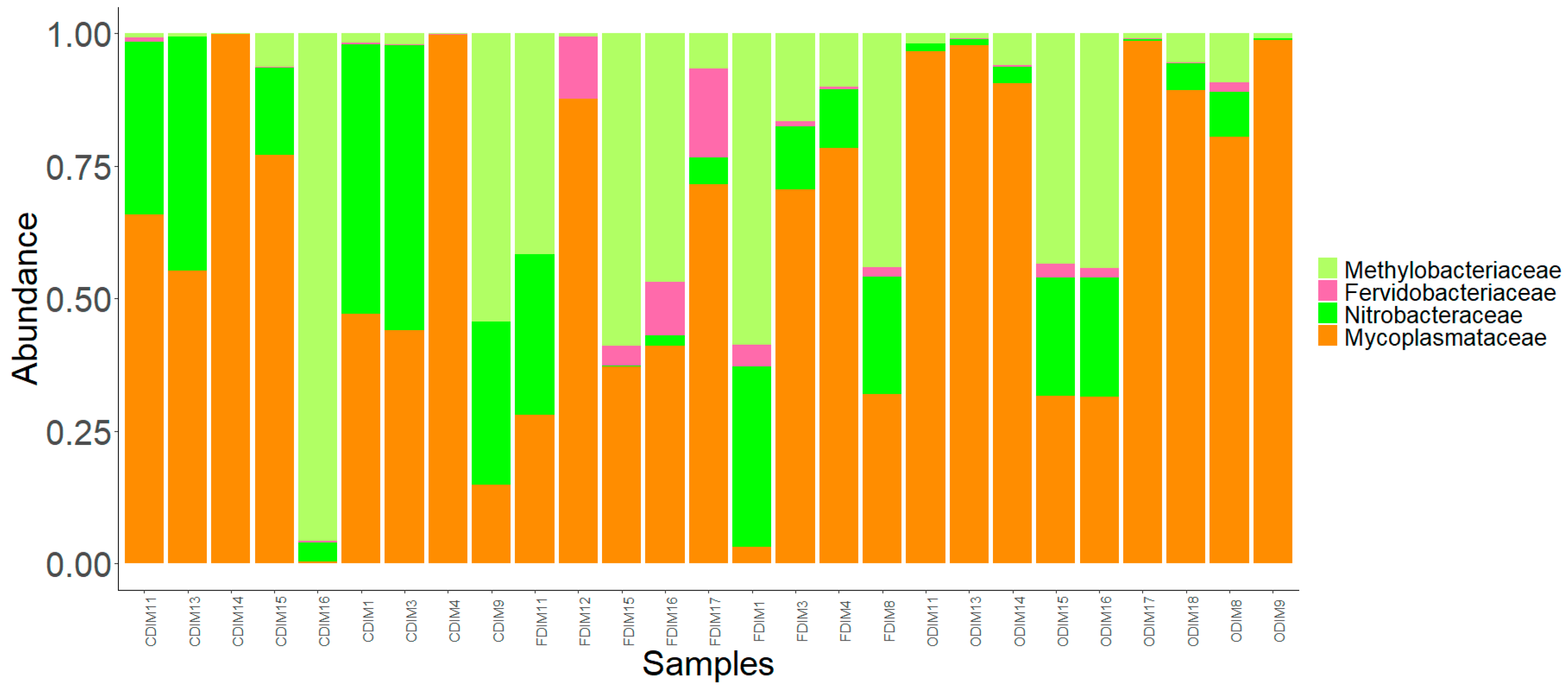
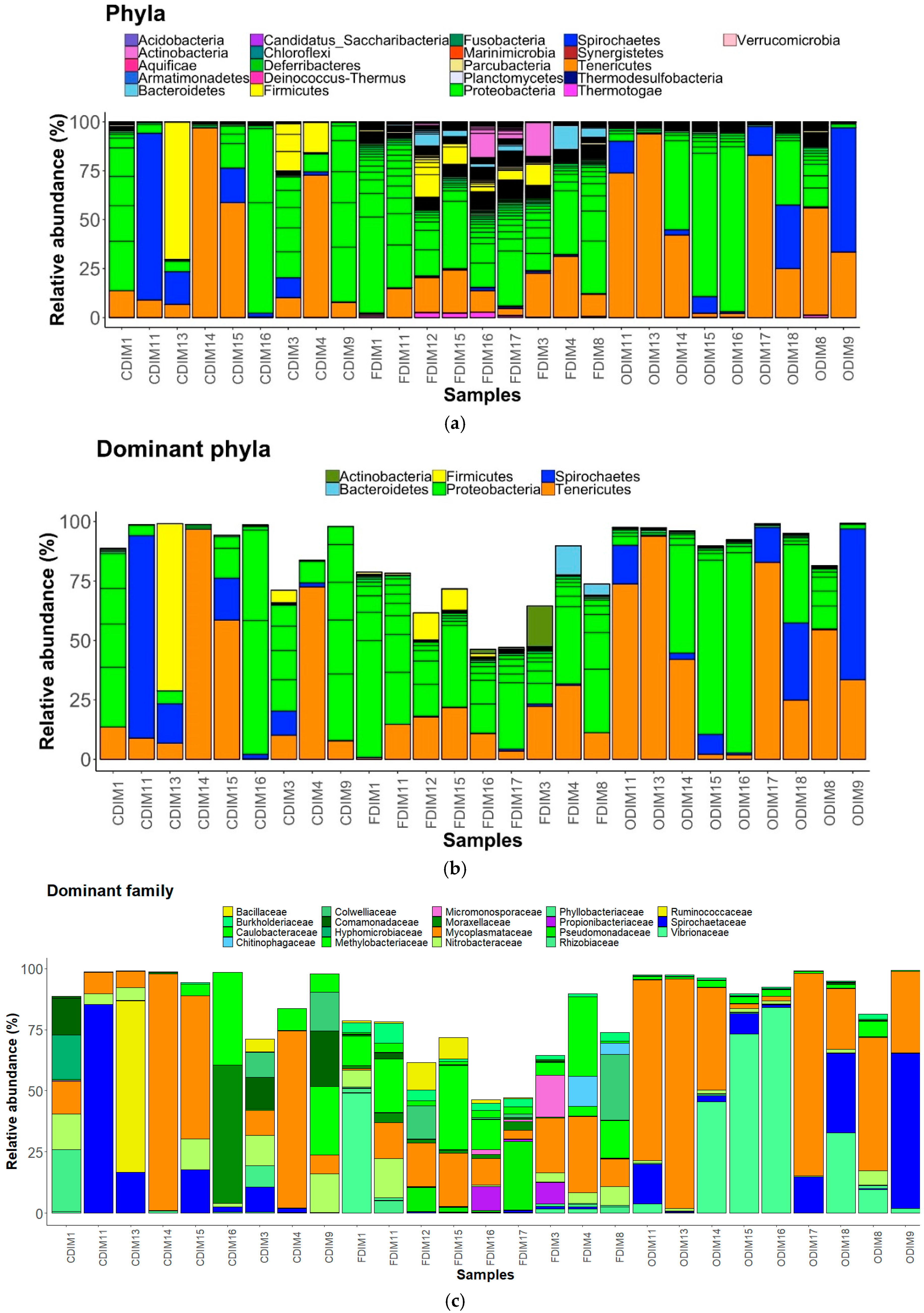
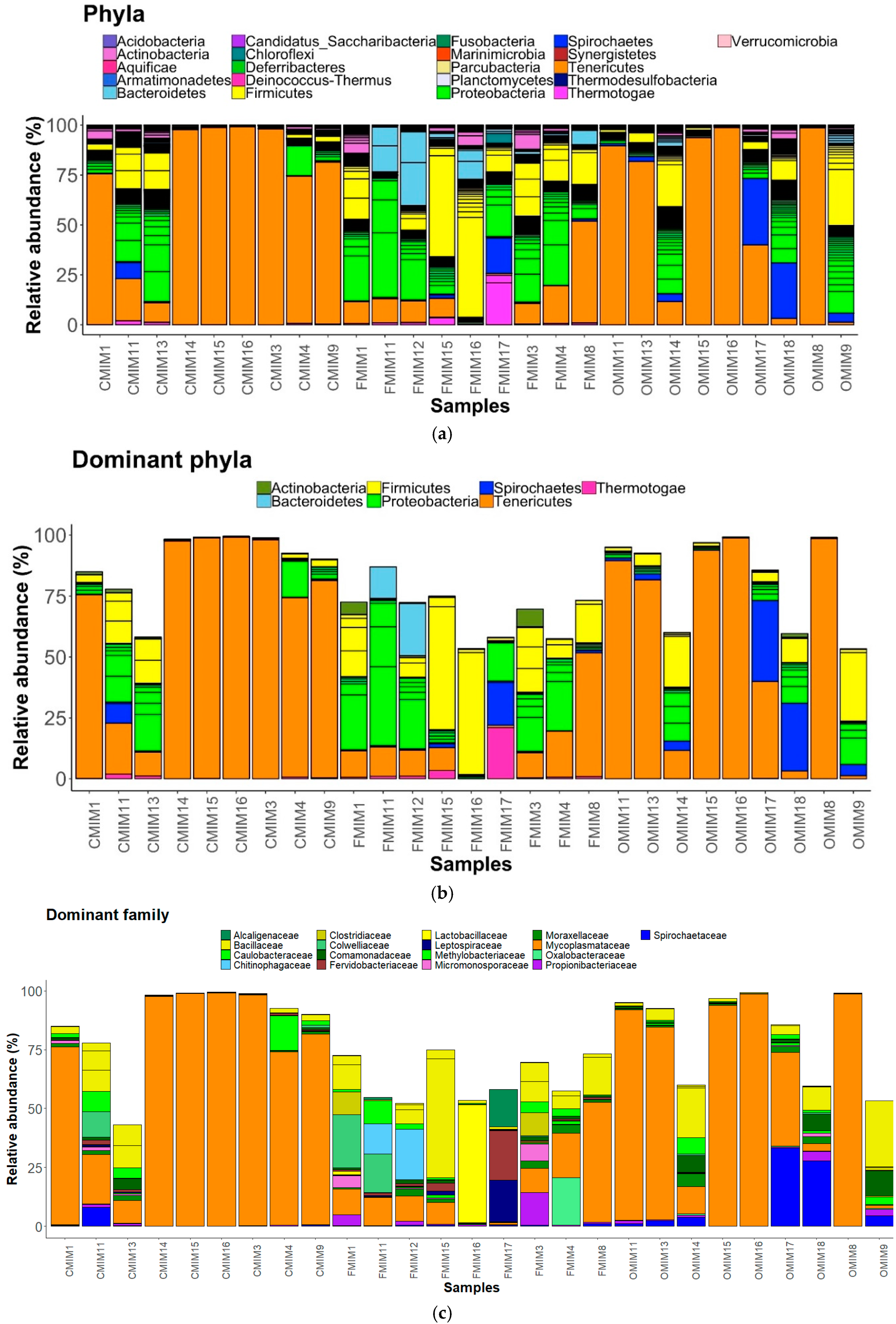
3.3. Changes in the Intestinal Mucus Bacterial Composition, Influenced by Antibiotics
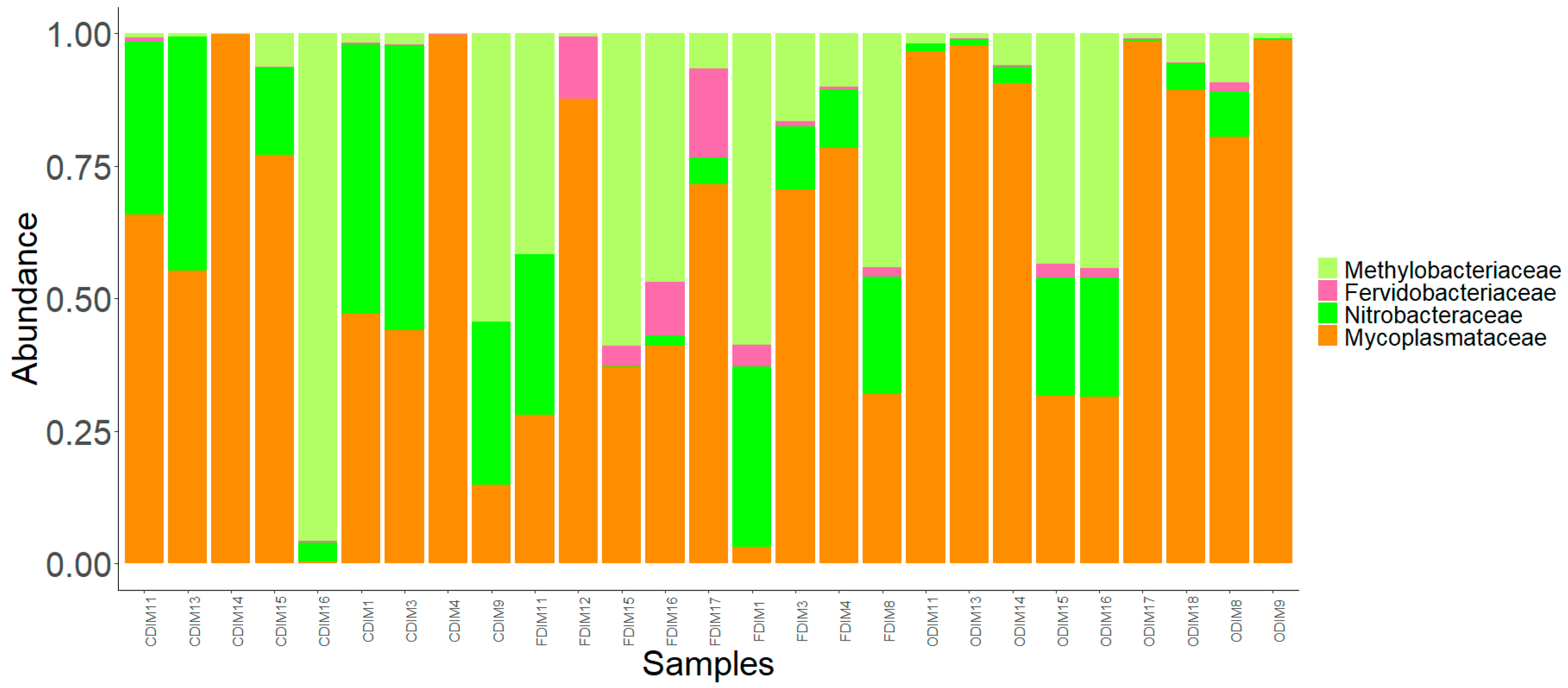
3.3.1. DI Mucus
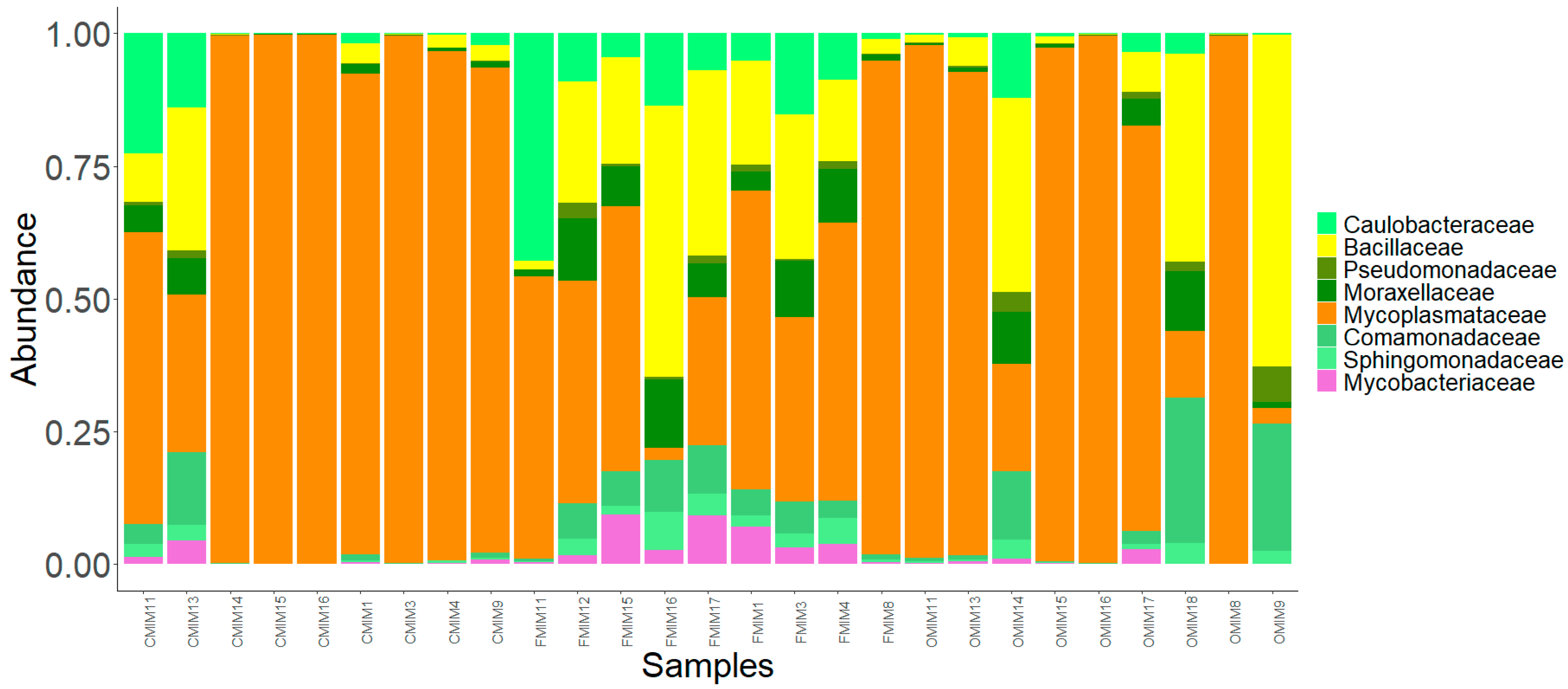
3.3.2. MI Mucus
3.4. Core Bacterial Communities of the Intestinal Mucus Microbiota
3.5. Significantly Abundant Taxa of the Intestinal Mucus Microbiota
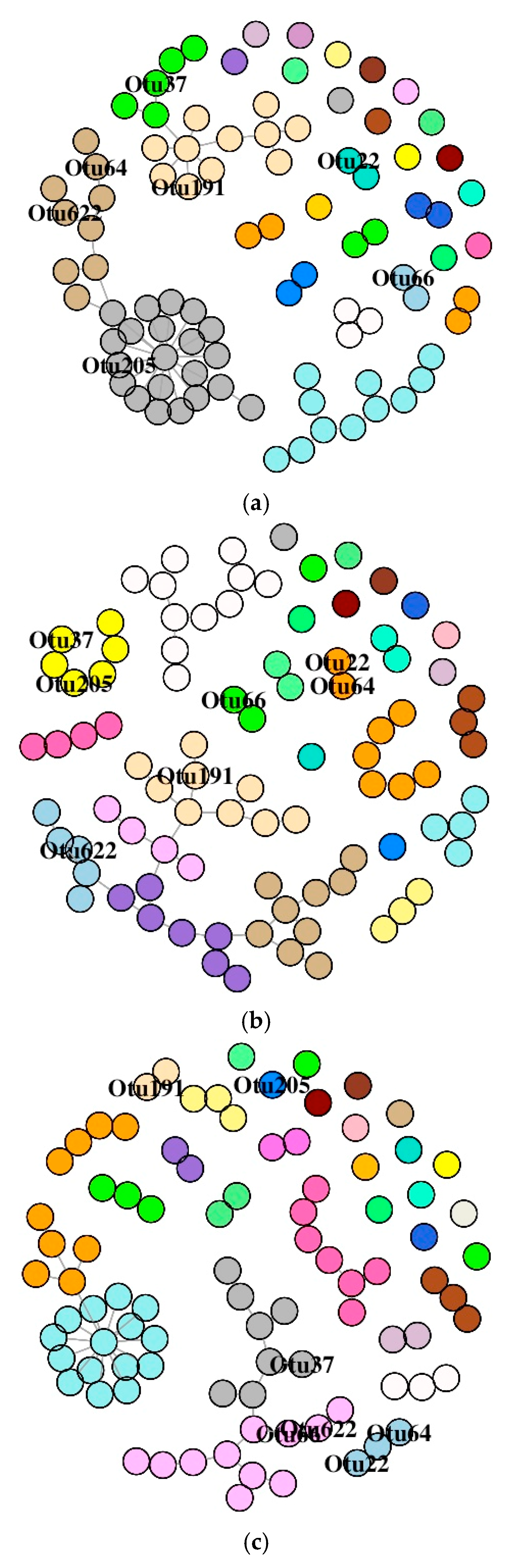
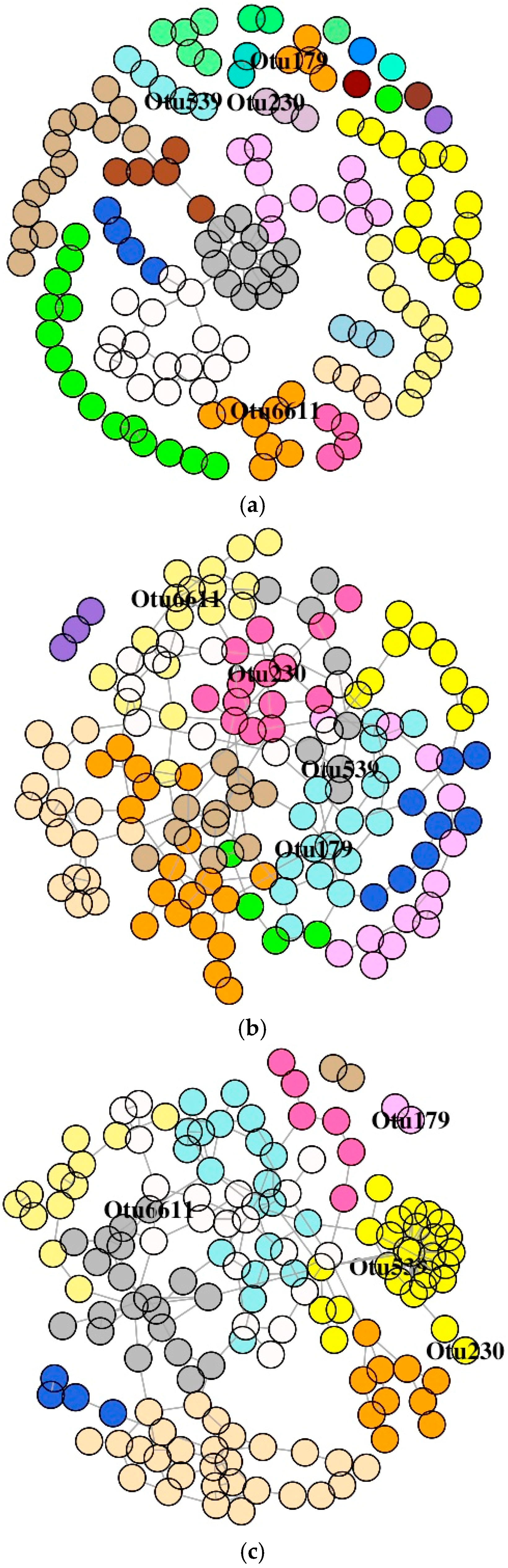
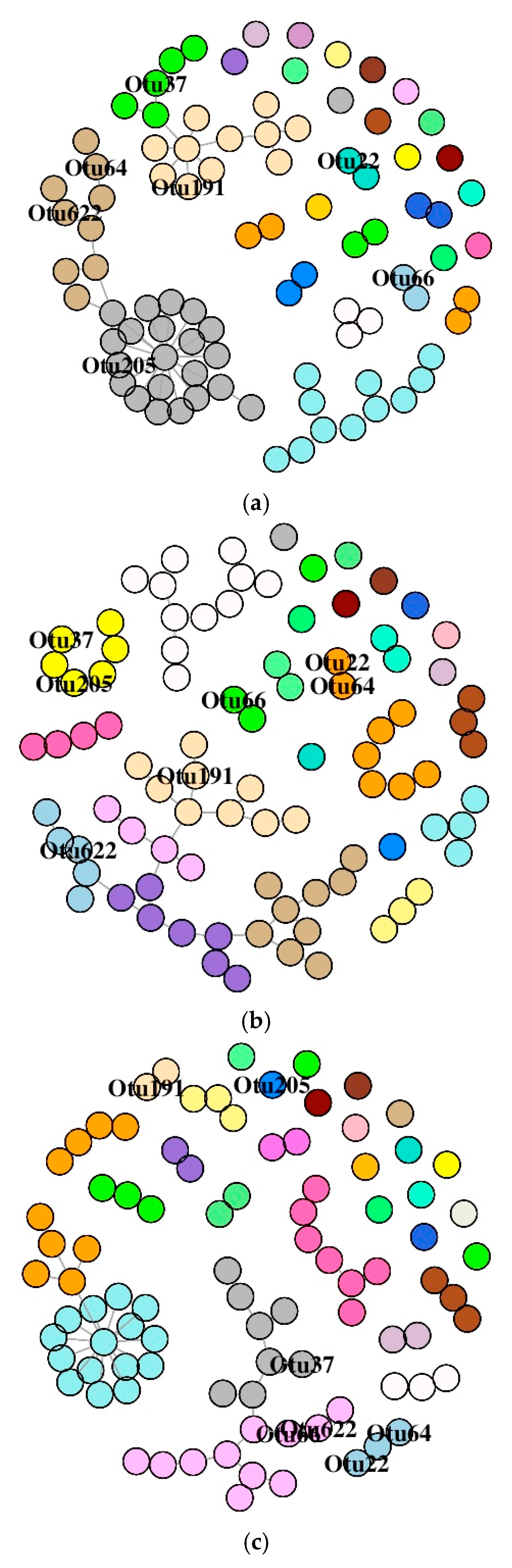
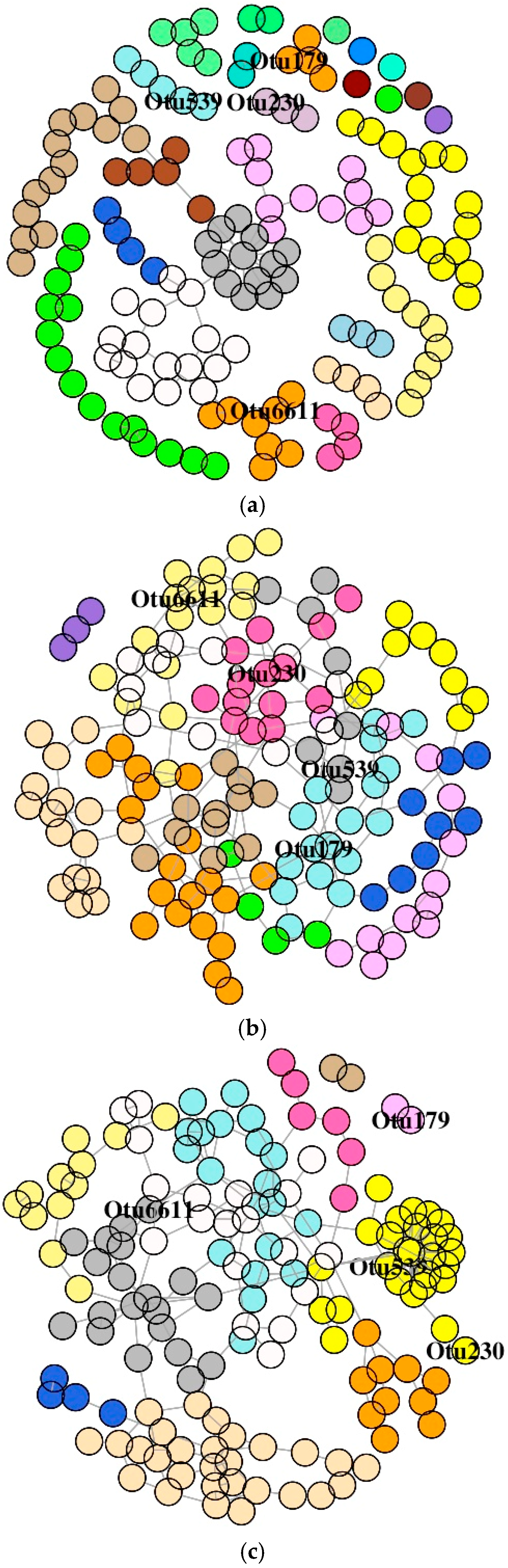
3.6. Co-Occurrence Network Description of OTUs
3.6.1. DI Mucus Bacteria
3.6.2. MI Mucus Bacteria
4. Discussion
4.1. Antibiotic Feeding Lifted the Richness and Diversity of the Intestinal Microbes
4.2. Antibiotic Feeding Altered the Composition of the Intestinal Mucus Microbial Consortia
4.3. Antibiotics Affected the Intestinal Mucus Microbial Association and Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Lederberg, J.; McCray, A.T. ‘Ome Sweet ’Omics a genealogical treasury of words. Scientist 2001, 15, 8. [Google Scholar]
- Senghor, B.; Sokhna, C.; Ruimy, R.; Lagier, J.-C. Gut microbiota diversity according to dietary habits and geographical provenance. Hum. Microbiome J. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Lawson, M.A.E.; Vaux, L.; Pin, C. Host-microbe interaction in the gastrointestinal tract. Environ. Microbiol. 2018, 20, 2337–2353. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M. Intestinal microbiota and its effects on the immune system. Cell Microbiol. 2014, 16, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Liu, W.; Piao, M.; Zhu, H. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids 2017, 49, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Semova, I.; Carten, J.D.; Stombaugh, J.; Mackey, L.C.; Knight, R.; Farber, S.A.; Rawls, J.F. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 2012, 12, 277–288. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, C.; Lofmark, S.; Edlund, C.; Jansson, J.K. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010, 156, 3216–3223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Zeng, M.Y.; Núñez, G. The interplay between host immune cells and gut microbiota in chronic inflammatory diseases. Exp. Mol. Med. 2017, 49, e339. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef] [PubMed]
- Okocha, R.C.; Olatoye, I.O.; Adedeji, O.B. Food safety impacts of antimicrobial use and their residues in aquaculture. Public Health Rev. 2018, 39, 21. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.D.; Godoy, F.A.; Lee, M.R. Current status of the use of antibiotics and the antimicrobial resistance in the Chilean salmon farms. Front. Microbiol. 2018, 9, 1284. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.E.M.; Schreier, H.J.; Lanska, L.; Hale, M.S. The rising tide of antimicrobial resistance in aquaculture: Sources, sinks and solutions. Mar. Drugs 2017, 15, 158. [Google Scholar] [CrossRef] [PubMed]
- Samuelsen, O.; Lunestad, B. Veterinary drug use in aquaculture. In Improving Farmed Fish Quality and Safety; Lie, Ø., Ed.; Woodhead Publishing: Cambridge, UK, 2008; pp. xiii–xvii. [Google Scholar] [CrossRef]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Bernatova, S.; Samek, O.; Pilat, Z.; Sery, M.; Jezek, J.; Jakl, P.; Siler, M.; Krzyzanek, V.; Zemanek, P.; Hola, V.; et al. Following the mechanisms of bacteriostatic versus bactericidal action using Raman spectroscopy. Molecules 2013, 18, 13188–13199. [Google Scholar] [CrossRef]
- Andriole, V.T. The quinolones: Past, present, and future. Clin. Infect. Dis. 2005, 41, S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Watson, D. Contaminants. In Food Chemical Safety; Watson, D.H., Ed.; Woodhead Publishing: Cambridge, UK, 2001; pp. 1–12. [Google Scholar] [CrossRef]
- Pankey, G.A.; Sabath, L.D. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of gram-positive bacterial infections. Clin. Infect. Dis. 2004, 38, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Llantén, S.; Vásquez-Ponce, F.; Barrientos-Espinoza, B.; Mardones, F.O.; Marshall, S.H.; Olivares-Pacheco, J. Extended antibiotic treatment in salmon farms select multiresistant gut bacteria with a high prevalence of antibiotic resistance genes. PLoS ONE 2018, 13, e0203641. [Google Scholar] [CrossRef] [PubMed]
- AS, F. Oxolinsyre vet. Skretting. Bredspektret Kjemoterapeutikum, ATCvet-nr.: QJ01M B91. Oslo, Norway.
- EMEA. Oxolinic Acid (Extension to Fish), 2nd ed.; The European Agency for the Evaluation of Medicinal Products: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Gupta, S.; Fečkaninová, A.; Lokesh, J.; Koščová, J.; Sørensen, M.; Fernandes, J.; Kiron, V. Lactobacillus dominate in the intestine of Atlantic salmon fed dietary probiotics. Front. Microbiol. 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the Miseq Illumina sequencing platform. J. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQc: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv 2016, 074161. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer-Verlag: New York, NY, USA, 2016; p. 259. [Google Scholar] [CrossRef]
- Lahti, L.; Shetty, S.; Blake, T.; Salojarvi, J. Microbiome R Package; Github: San Francisco, CA, USA, 2017. [Google Scholar]
- Jost, L. Entropy and diversity. Oikos 2006, 113, 363–375. [Google Scholar] [CrossRef]
- Fukuyama, J.; McMurdie, P.J.; Dethlefsen, L.; Relman, D.A.; Holmes, S. Comparisons of distance methods for combining covariates and abundances in microbiome studies. Pac. Symp. Biocomput. 2012, 213–224. [Google Scholar]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, Z.; Mueller, C.; Miraldi, E.; Bonneau, R. SpiecEasi: Sparse Inverse Covariance for Ecological Statistical Inference, R package version 1.0.2.; 2018. [Google Scholar]
- Raymann, K.; Bobay, L.-M.; Moran, N.A. Antibiotics reduce genetic diversity of core species in the honeybee gut microbiome. Mol. Ecol. 2018, 27, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J. Antibiotic use and its consequences for the normal microbiome. Science 2016, 352, 544–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Limenitakis, J.P.; Fuhrer, T.; Geuking, M.B.; Lawson, M.A.; Wyss, M.; Brugiroux, S.; Keller, I.; Macpherson, J.A.; Rupp, S.; et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun. 2015, 6, 8292. [Google Scholar] [CrossRef] [PubMed]
- Zarepour, M.; Bhullar, K.; Montero, M.; Ma, C.; Huang, T.; Velcich, A.; Xia, L.; Vallance, B.A. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect. Immun. 2013, 81, 3672–3683. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, H.; Shi, Q.; Wang, N.; Zhang, Z.; Xiong, C.; Liu, J.; Chen, Y.; Jiang, L.; Jiang, Q. Effects of oral florfenicol and azithromycin on gut microbiota and adipogenesis in mice. PLoS ONE 2017, 12, e0181690. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef]
- Wang, E.; Yuan, Z.; Wang, K.; Gao, D.; Liu, Z.; Liles, M.R. Consumption of florfenicol-medicated feed alters the composition of the channel catfish intestinal microbiota including enriching the relative abundance of opportunistic pathogens. Aquaculture 2019, 501, 111–118. [Google Scholar] [CrossRef]
- Marker, L.M.; Hammer, A.S.; Andresen, L.; Isaack, P.; Clausen, T.; Byskov, K.; Honoré, O.L.; Jensen, S.K.; Bahl, M.I. Short-term effect of oral amoxicillin treatment on the gut microbial community composition in farm mink (Neovison vison). FEMS Microbiol. Ecol. 2017, 93, fix092. [Google Scholar] [CrossRef] [PubMed]
- Bohn, K.; Pavlick, R.; Reu, B.; Kleidon, A. The strengths of r- and K-selection shape diversity-disturbance relationships. PLoS ONE 2014, 9, e95659. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Savage, V.M.; Yeh, P.J. Intermediate levels of antibiotics may increase diversity of colony size phenotype in bacteria. Comput. Struct. Biotechnol. J. 2018, 16, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cobas, A.E.; Artacho, A.; Knecht, H.; Ferrús, M.L.; Friedrichs, A.; Ott, S.J.; Moya, A.; Latorre, A.; Gosalbes, M.J. Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS ONE 2013, 8, e80201. [Google Scholar] [CrossRef] [PubMed]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- Gajardo, K.; Rodiles, A.; Kortner, T.M.; Krogdahl, Å.; Bakke, A.M.; Merrifield, D.L.; Sørum, H. A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): A basis for comparative gut microbial research. Sci. Rep. 2016, 6, 30893. [Google Scholar] [CrossRef]
- Gupta, S.; Lokesh, J.; Abdelhafiz, Y.A.; Pierre, R.; Sørensen, M.; Fernandes, J.; Kiron, V. Macroalga-derived alginate oligosaccharide alters certain intestinal bacteria of Atlantic salmon. Front. Microbiol. 2018. Submitted. [Google Scholar]
- Romero, J.; Einar, R.; Merrifield, D.L. The gut microbiota of fish. In Aquaculture Nutrition: Gut Health, Probiotics and Prebiotics, 1st ed.; Merrifield, D., Ringø, E., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2014; p. 488. [Google Scholar]
- Vikram, S.; Guerrero, L.D.; Makhalanyane, T.P.; Le, P.T.; Seely, M.; Cowan, D.A. Metagenomic analysis provides insights into functional capacity in a hyperarid desert soil niche community. Environ. Microbiol. 2016, 18, 1875–1888. [Google Scholar] [CrossRef]
- Austin, B.; Austin, D. Pathogenicity. In Bacterial Fish Pathogens; Diseases of Farmed and Wild Fish, 4th ed.; Praxis Publishing: Chichester, UK, 2007. [Google Scholar]
- Schmidt, V.T.; Reveillaud, J.; Zettler, E.; Mincer, T.J.; Murphy, L.; Amaral-Zettler, L.A. Oligotyping reveals community level habitat selection within the genus Vibrio. Front. Microbiol. 2014, 5, 563. [Google Scholar] [CrossRef]
- Schmidt, V.; Gomez-Chiarri, M.; Roy, C.; Smith, K.; Amaral-Zettler, L. Subtle microbiome manipulation using probiotics reduces antibiotic-associated mortality in fish. Msystems 2017, 2, e00133-17. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.R.; Scott, E.J., 2nd; Dyer, D.W. Whole-genome phylogenies of the family Bacillaceae and expansion of the sigma factor gene family in the Bacillus cereus species-group. BMC Genom. 2011, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Kiron, V. Gastrointestinal microorganisms of fish and probiotics. In Dietary Nutrients, Additives, and Fish Health; Lee, C.S., Lim, C.D.M.G., III, Webster, C.D., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2015; pp. 283–304. [Google Scholar] [CrossRef]
- Picardeau, M. The Family Leptospiraceae. In the Prokaryotes: Other Major Lineages of Bacteria and the Archaea; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 711–729. [Google Scholar] [CrossRef]
- Mgode, G.F.; Mhamphi, G.G.; Katkweba, A.S.; Thomas, M. Leptospira infections in freshwater fish in Morogoro Tanzania: A hidden public health threat. Tanzan. J. Health Res. 2014, 16, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Davies, S.J.; Waines, P.; Emery, M.; Castex, M.; Gioacchini, G.; Carnevali, O.; Bickerdike, R.; Romero, J.; Merrifield, D.L. Dietary synbiotic application modulates Atlantic salmon (Salmo salar) intestinal microbial communities and intestinal immunity. Fish Shellfish Immunol. 2013, 35, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, M.S.; McGinnity, P.; Dionne, M.; Letourneau, J.; Thonier, F.; Carvalho, G.R.; Creer, S.; Derome, N. The biogeography of the Atlantic salmon (Salmo salar) gut microbiome. ISME J. 2016, 10, 1280–1284. [Google Scholar] [CrossRef]
- Yoon, M.Y.; Yoon, S.S. Disruption of the gut ecosystem by antibiotics. Yonsei Med. J. 2018, 59, 4–12. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef]
- McNally, L.; Brown, S.P. Microbiome: Ecology of stable gut communities. Nat. Microbiol. 2016, 1, 15016. [Google Scholar] [CrossRef]
- Carareto Alves, L.M.; de Souza, J.A.M.; Varani, A.d.M.; Lemos, E.G.d.M. The family Rhizobiaceae. In the Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 419–437. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alpha Diversity | Distal Intestine | Mid Intestine | ||||||
|---|---|---|---|---|---|---|---|---|
| Groups | p-Value | Groups | Mean ± SD | Groups | p-Value | Groups | Mean ± SD | |
| Species richness | FDM-CDM | 0.000 | CDM | 55.11 ± 19.23 a | FMM-CMM | 0.223 | CMM | 120.78 ± 59.28 a,b |
| ODM-CDM | 0.104 | FDM | 215.22 ± 59.11 b | OMM-CMM | 1.000 | FMM | 168.44 ± 37.71 b | |
| ODM-FDM | 0.130 | ODM | 120.67 ± 55.65 a,b | OMM-FMM | 0.048 | OMM | 109.44 ± 38.31 a | |
| Shannon diversity | FDM-CDM | 0.001 | CDM | 4.51 ± 3.52 a | FMM-CMM | 1.121 | CMM | 1.22 ± 1.28 |
| ODM-CDM | 1.000 | FDM | 21.38 ± 12.22 b | OMM-CMM | 1.000 | FMM | 2.68 ± 0.43 | |
| ODM-FDM | 0.001 | ODM | 3.68 ± 2.36 a,b | OMM-FMM | 0.462 | OMM | 1.62 ± 1.41 | |
| Simpson diversity | FDM-CDM | 0.020 | CDM | 3.61 ± 3.06 a | FMM-CMM | 0.108 | CMM | 3.80 ± 4.99 |
| ODM-CDM | 1.000 | FDM | 8.90 ± 4.19 b | OMM-CMM | 1.000 | FMM | 7.48 ± 3.77 | |
| ODM-FDM | 0.001 | ODM | 2.10 ± 0.85 a,b | OMM-FMM | 0.297 | OMM | 4.69 ± 4.80 | |
| PD | FDM-CDM | 0.001 | CDM | 183.05 ± 46.57 a | FMM-CMM | 0.112 | CMM | 312.29 ± 108.90 a |
| ODM-CDM | 0.121 | FDM | 478.74 ± 99.51 b | OMM-CMM | 1.000 | FMM | 400.98 ± 57.80 a,b | |
| ODM-FDM | 0.121 | ODM | 319.22 ± 105.72 a,b | OMM-FMM | 0.066 | OMM | 291.83 ± 72.38 a,c | |
| Groups | Control | F-Fed Group | O-Fed Group | |||
|---|---|---|---|---|---|---|
| Sample Type | DI | MI | DI | MI | DI | MI |
| Phyla | ||||||
| Proteobacteria | 41.64 ± 40.32 | 14.99 ± 19.44 | 61.01 ± 17.98 | 33.71 ± 18.32 | 37.23 ± 36.10 | 17.75 ± 19.26 |
| Bacteroidetes | 0.06 ± 0.05 | 0.98 ± 0.97 | 5.73 ± 4.23 | 11.04 ± 12.66 | 0.55 ± 0.54 | 2.23 ± 2.57 |
| Tenericutes | 30.55 ± 35.59 | 72.78 ± 34.25 | 14.91 ± 9.58 | 13.80 ± 14.97 | 45.48 ± 33.53 | 57.63 ± 43.13 |
| Firmicutes | 12.45 ± 23.38 | 6.87 ± 9.91 | 10.05 ± 7.95 | 28.86 ± 22.03 | 0.67 ± 0.86 | 11.73 ± 14.71 |
| Actinobacteria | 0.25 ± 0.38 | 2.41 ± 2.71 | 5.62 ± 7.37 | 5.16 ± 5.34 | 0.13 ± 0.15 | 2.28 ± 2.39 |
| Spirochaetes | 14.90 ± 27.33 | 1.20 ± 2.80 | 0.92 ± 0.53 | 2.74 ± 5.94 | 15.60 ± 20.83 | 8.17 ± 12.89 |
| Thermotogae | - | 0.54 ± 0.72 | - | 3.75 ± 7.96 | -; | 0.03 ± 0.05 |
| Family | ||||||
| Mycoplasmataceae | 30.55 ± 35.59 | 72.78 ± 34.26 | 14.92 ± 9.57 | 13.80 ± 14.97 | 45.48 ± 33.52 | 57.63 ± 43.13 |
| Comamonadaceae | 5.77 ± 8.97 | 2.95 ± 7.04 | 1.35 ± 0.83 | 2.47 ± 4.28 | 0.19 ± 0.21 | 3.76 ± 4.34 |
| Bacillaceae | 0.71 ± 1.74 | 5.79 ± 8.75 | 4.87 ± 5.77 | 14.30 ± 16.75 | 0.05 ± 0.07 | 9.59 ± 12.02 |
| Sphingomonadaceae | 0.00 ± 0.01 | 0.51 ± 0.58 | 1.09 ± 1.11 | 0.85 ± 0.94 | 0.16 ± 0.17 | 0.86 ± 0.94 |
| Moraxellaceae | 6.39 ± 18.77 | 1.00 ± 1.06 | 2.17 ± 1.39 | 1.63 ± 1.42 | 0.24 ± 0.33 | 2.82 ± 3.67 |
| Mycobacteriaceae | 0.00 ± 0.00 | 0.40 ± 0.51 | 0.39 ± 0.40 | 0.73 ± 0.64 | 0.01 ± 0.27 | 0.35 ± 0.50 |
| Caulobacteraceae | 1.84 ± 3.66 | 1.90 ± 2.92 | 5.23 ± 10.30 | 2.50 ± 3.03 | 0.13 ± 0.11 | 1.26 ± 2.18 |
| Pseudomonadaceae | 0.26 ± 0.45 | 0.25 ± 0.29 | 6.00 ± 9.41 | 2.44 ± 2.06 | 0.19 ± 0.28 | 1.33 ± 1.54 |
| Alcaligenaceae | 0.00 ± 0.00 | 0.04 ± 0.11 | 0.15 ± 0.40 | 1.87 ± 5.18 | 0.00 ± 0.00 | 0.04 ± 0.12 |
| Chitinophagaceae | 0.00 ± 0.00 | 0.08 ± 0.15 | 1.88 ± 4.16 | 3.83 ± 7.85 | 0.02 ± 0.02 | 0.17 ± 0.46 |
| Clostridiaceae | 1.11 ± 3.33 | 0.19 ± 0.22 | 1.08 ± 1.75 | 3.22 ± 4.67 | 0.00 ± 0.00 | 0.36 ± 0.85 |
| Colwelliaceae | 2.95 ± 5.91 | 1.67 ± 4.49 | 4.78 ± 9.32 | 4.41 ± 8.68 | 0.01 ± 0.02 | 0.27 ± 0.36 |
| Fervidobacteriaceae | 0.07 ± 0.04 | 0.51 ± 0.70 | 1.16 ± 1.06 | 3.56 ± 7.58 | 0.19 ± 0.35 | 0.03 ± 0.05 |
| Lactobacillaceae | 0.01 ± 0.01 | 0.18 ± 0.17 | 0.83 ± 0.46 | 6.19 ± 16.51 | 0.29 ± 0.38 | 0.29 ± 0.43 |
| Leptospiraceae | 0.05 ± 0.06 | 0.22 ± 0.29 | 0.71 ± 0.59 | 3.46 ± 8.44 | 0.10 ± 0.16 | 0.01 ± 0.02 |
| Methylobacteriaceae | 8.00 ± 14.43 | 1.77 ± 4.90 | 11.77 ± 11.13 | 0.49 ± 0.48 | 2.15 ± 1.79 | 0.92 ± 1.13 |
| Micromonosporaceae | 0.03 ± 0.08 | 0.38 ± 0.54 | 2.25 ± 5.61 | 1.54 ± 2.66 | 0.00 ± 0.01 | 0.26 ± 0.48 |
| Oxalobacteraceae | 0.00 ± 0.00 | 0.13 ± 0.26 | 1.20 ± 2.56 | 2.27 ± 6.76 | 0.02 ± 0.04 | 0.24 ± 0.23 |
| Propionibacteriaceae | 0.00 ± 0.01 | 0.37 ± 0.44 | 2.26 ± 4.04 | 2.57 ± 4.52 | 0.02 ± 0.01 | 1.08 ± 1.32 |
| Spirochaetaceae | 14.88 ± 27.32 | 1.04 ± 2.64 | 0.51 ± 0.38 | 0.43 ± 0.35 | 15.56 ± 20.85 | 8.16 ± 12.89 |
| Vibrionaceae | 1.18 ± 2.36 | ND | 6.78 ± 17.12 | ND | 29.08 ± 34.57 | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Fernandes, J.; Kiron, V. Antibiotic-Induced Perturbations Are Manifested in the Dominant Intestinal Bacterial Phyla of Atlantic Salmon. Microorganisms 2019, 7, 233. https://doi.org/10.3390/microorganisms7080233
Gupta S, Fernandes J, Kiron V. Antibiotic-Induced Perturbations Are Manifested in the Dominant Intestinal Bacterial Phyla of Atlantic Salmon. Microorganisms. 2019; 7(8):233. https://doi.org/10.3390/microorganisms7080233
Chicago/Turabian StyleGupta, Shruti, Jorge Fernandes, and Viswanath Kiron. 2019. "Antibiotic-Induced Perturbations Are Manifested in the Dominant Intestinal Bacterial Phyla of Atlantic Salmon" Microorganisms 7, no. 8: 233. https://doi.org/10.3390/microorganisms7080233
APA StyleGupta, S., Fernandes, J., & Kiron, V. (2019). Antibiotic-Induced Perturbations Are Manifested in the Dominant Intestinal Bacterial Phyla of Atlantic Salmon. Microorganisms, 7(8), 233. https://doi.org/10.3390/microorganisms7080233