VelA and LaeA are Key Regulators of Epichloë festucae Transcriptomic Response during Symbiosis with Perennial Ryegrass


Abstract
:1. Background
2. Results
2.1. Choosing the Conditions for Transcriptomic Studies
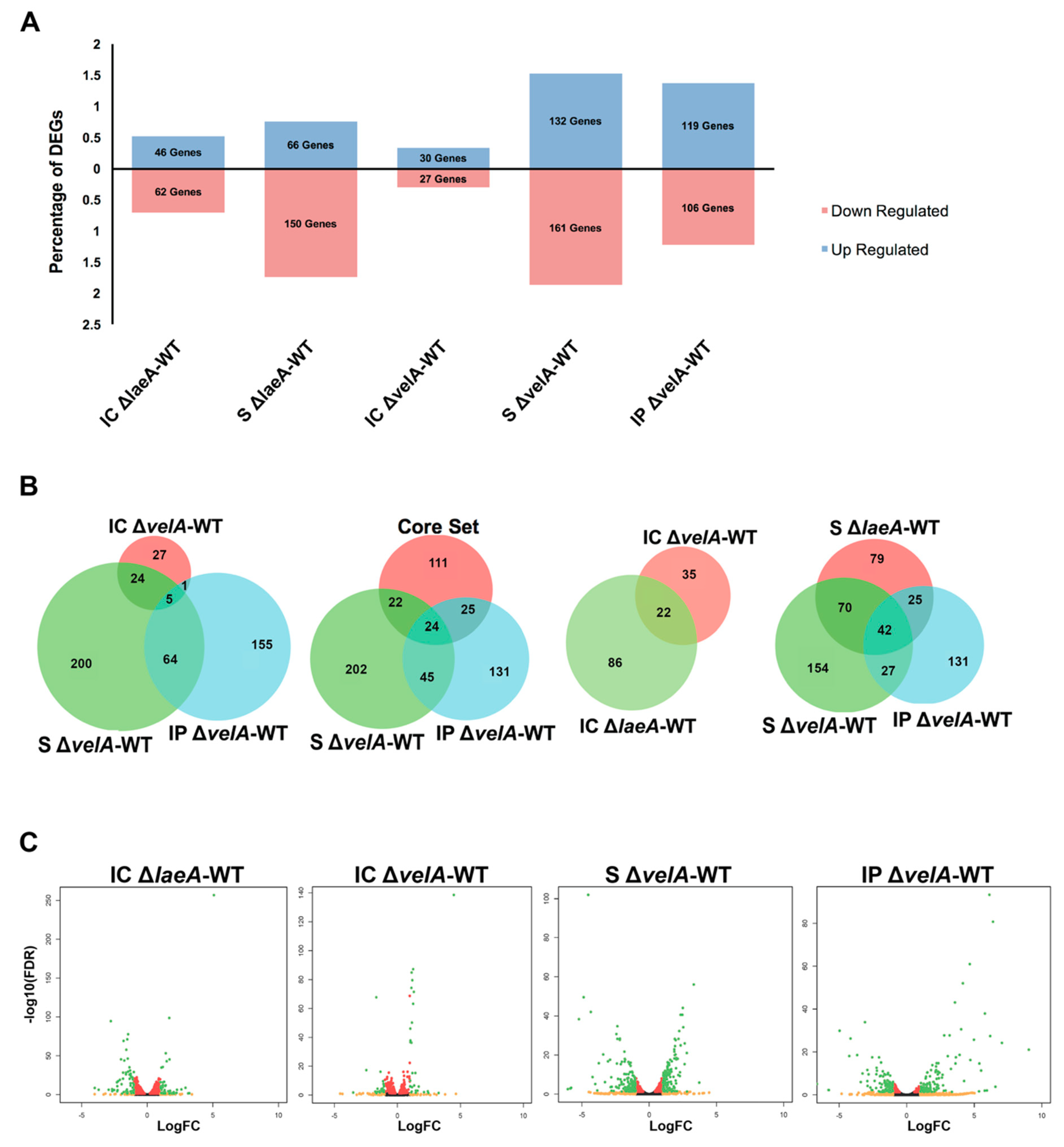
2.2. General Description of RNA Sequencing Results
2.3. Gene Ontology (GO) Enrichment Analysis on DEGs
2.4. Functional Annotations of Differentially Expressed E. Festucae Genes
2.4.1. Changes in the Expression of Genes Encoding Orthologues of Velvet Family Members
2.4.2. DEGs in Different Functional Categories
2.4.3. Changes in the Expression of Genes Encoding Membrane Transporters
2.4.4. Changes in the Expression of Genes Encoding Enzymes with Host Cell Wall-Degrading Activity
2.4.5. Changes in the Expression of Genes Encoding Proteins Involved in Fungal Cell Wall Composition
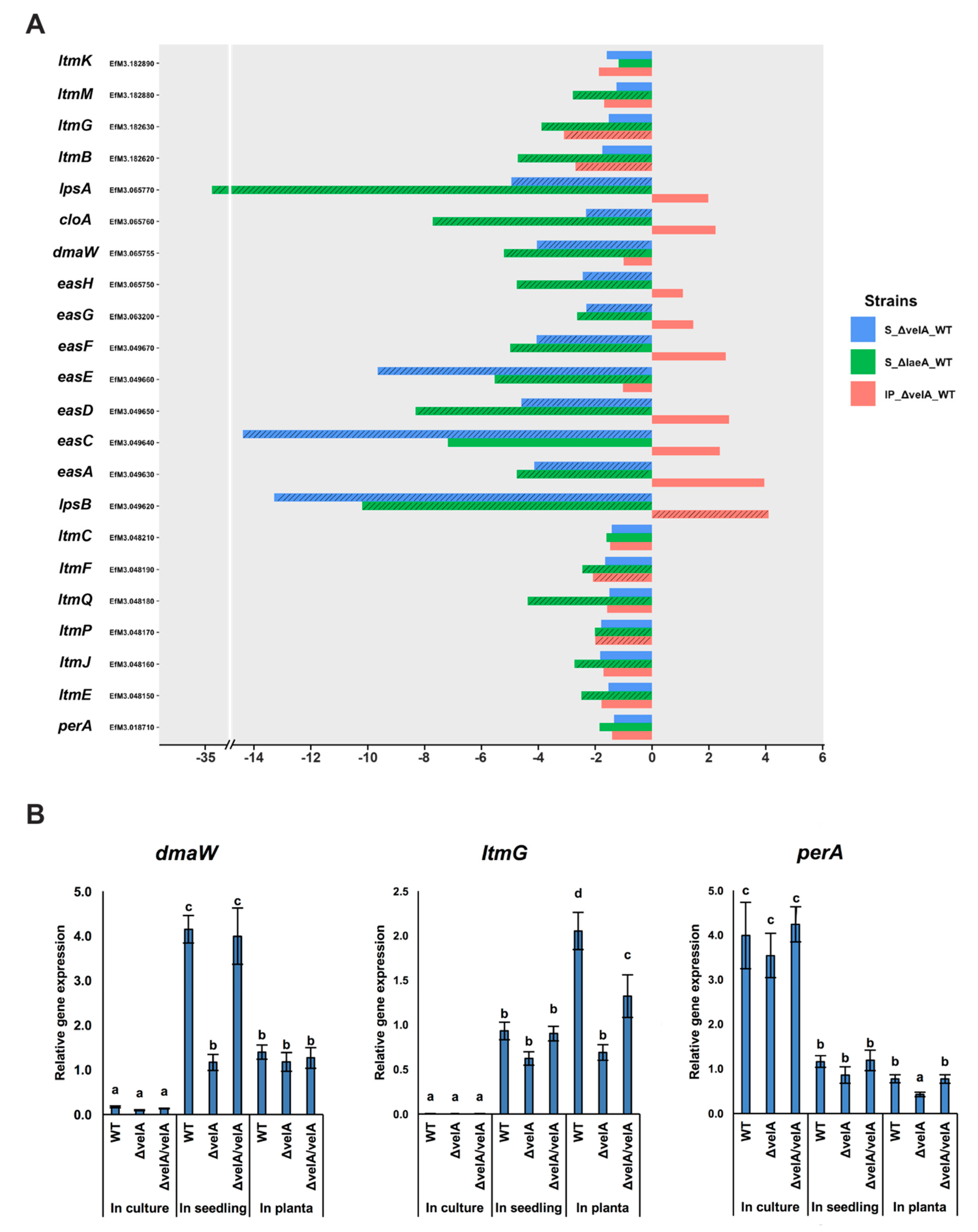
2.4.6. VelA is Required for Secondary Metabolite Gene Expression and Production
2.4.7. Changes in the Expression of Genes Encoding Putative Small Secreted Proteins (SSPs)
3. Discussion
4. Methods
4.1. Sample Preparation
4.2. RNA Extraction and Quantitative Real-Time RT-PCR Analysis
4.3. HiSeq Results’ Analysis
4.4. General Bioinformatics Analyses
4.5. Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leuchtmann, A.; Schardl, C.L.; Siegel, M.R. Sexual Compatibility and Taxonomy of a New Species of Epichloe Symbiotic with Fine Fescue Grasses. Mycologia 1994, 86, 802–812. [Google Scholar]
- Christensen, M.J.; Bennett, R.J.; Schmid, J. Growth of Epichloë/Neotyphodium and p-endophytes in leaves of Lolium and Festuca grasses. Mycol. Res. 2002, 106, 93–106. [Google Scholar] [CrossRef]
- Schardl, C.L. The Epichloae, Symbionts of the Grass Subfamily Poöideae 1. Ann. Mo. Bot. Gard. 2010, 97, 646–665. [Google Scholar] [CrossRef]
- Johnson, L.J.; De Bonth, A.C.M.; Briggs, L.R.; Caradus, J.R.; Finch, S.C.; Fleetwood, D.J.; Fletcher, L.R.; Hume, D.E.; Johnson, R.D.; Popay, A.J.; et al. The exploitation of epichloae endophytes for agricultural benefit. Fungal Divers. 2013, 60, 171–188. [Google Scholar] [CrossRef]
- Arachevaleta, M.; Bacon, C.W.; Hoveland, C.S.; Radcliffe, D.E. Effect of the Tall Fescue Endophyte on Plant Response to Environmental Stress. Agron. J. 1989, 81, 83–90. [Google Scholar] [CrossRef]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-Symbiotic Fungi as Chemical Engineers: Multi-Genome Analysis of the Clavicipitaceae Reveals Dynamics of Alkaloid Loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.J.; Bennett, R.J.; Ansari, H.A.; Koga, H.; Johnson, R.D.; Bryan, G.T.; Simpson, W.R.; Koolaard, J.P.; Nickless, E.M.; Voisey, C.R. Epichloë endophytes grow by intercalary hyphal extension in elongating grass leaves. Fungal Genet. Biol. 2008, 45, 84–93. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Spiering, M.J.; Scott, V.; Lane, G.A.; Christensen, M.J.; Schmid, J. In Planta Regulation of Extension of an Endophytic Fungus and Maintenance of High Metabolic Rates in Its Mycelium in the Absence of Apical Extension. Appl. Environ. Microbiol. 2001, 67, 5377–5383. [Google Scholar] [CrossRef] [Green Version]
- Schmid, J.; Day, R.; Zhang, N.; Dupont, P.-Y.; Cox, M.P.; Schardl, C.L.; Minards, N.; Truglio, M.; Moore, N.; Harris, D.R.; et al. Host Tissue Environment Directs Activities of an Epichloë Endophyte, While It Induces Systemic Hormone and Defense Responses in Its Native Perennial Ryegrass Host. Mol. Plant-Microbe Interact. 2016, 30, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Charlton, N.D.; Shoji, J.-Y.; Ghimire, S.R.; Nakashima, J.; Craven, K.D. Deletion of the Fungal Gene soft Disrupts Mutualistic Symbiosis between the Grass Endophyte Epichloë festucae and the Host Plant. Eukaryot. Cell 2012, 11, 1463–1471. [Google Scholar] [CrossRef] [Green Version]
- Rahnama, M.; Johnson, R.; Voisey, C.R.; Simpson, W.R.; Fleetwood, D.J. The Global Regulatory Protein VelA Is Required for Symbiosis Between the Endophytic Fungus Epichloë festucae and Lolium perenne. Mol. Plant-Microbe Interact. 2018, 31, 591–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahnama, M.; MacLean, P.; Fleetwood, D.; Johnson, R. The LaeA orthologue in Epichloë festucae is required for symbiotic interaction with Lolium perenne. Fungal Genet. Biol. 2019, 129, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Christensen, M.J.; Takemoto, D.; Park, P.; Scott, B. Reactive Oxygen Species Play a Role in Regulating a Fungus–Perennial Ryegrass Mutualistic Interaction. Plant Cell 2006, 18, 1052–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyon, G.-S.; Tanaka, A.; Takemoto, D.; Park, P.; Scott, B. NoxA activation by the small GTPase RacA is required to maintain a mutualistic symbiotic association between Epichloë festucae and perennial ryegrass. Mol. Microbiol. 2008, 68, 1165–1178. [Google Scholar]
- Takemoto, D.; Kamakura, S.; Saikia, S.; Becker, Y.; Wrenn, R.; Tanaka, A.; Sumimoto, H.; Scott, B. Polarity proteins Bem1 and Cdc24 are components of the filamentous fungal NADPH oxidase complex. Proc. Natl. Acad. Sci. USA 2011, 108, 2861–2866. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Cartwright, G.M.; Saikia, S.; Kayano, Y.; Takemoto, D.; Kato, M.; Tsuge, T.; Scott, B. ProA, a transcriptional regulator of fungal fruiting body development, regulates leaf hyphal network development in the Epichloë festucae–Lolium perenne symbiosis. Mol. Microbiol. 2013, 90, 551–568. [Google Scholar] [CrossRef]
- Johnson, L.J.; Koulman, A.; Christensen, M.; Lane, G.A.; Fraser, K.; Forester, N.; Johnson, R.D.; Bryan, G.T.; Rasmussen, S. An Extracellular Siderophore Is Required to Maintain the Mutualistic Interaction of Epichloë festucae with Lolium perenne. PLoS Pathog. 2013, 9, e1003332. [Google Scholar] [CrossRef] [Green Version]
- Forester, N.T.; Lane, G.A.; Steringa, M.; Lamont, I.L.; Johnson, L.J. Contrasting roles of fungal siderophores in maintaining iron homeostasis in Epichloë festucae. Fungal Genet. Biol. 2018, 111, 60–72. [Google Scholar] [CrossRef]
- Becker, Y.; Brasell, E.; Becker, M.; Hassing, B.; Reinhold, L.; Scott, B.; Eaton, C.J.; May, K.J.; Cartwright, G.M. The Fungal Cell-Wall Integrity MAPK Cascade Is Crucial for Hyphal Network Formation and Maintenance of Restrictive Growth of Epichloë festucae in Symbiosis With Lolium perenne. Mol. Plant-Microbe Interact. 2015, 28, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Dupont, P.-Y.; Solomon, P.S.; Clayton, W.; Scott, B.; Eaton, C.J.; Cox, M.P. A Core Gene Set Describes the Molecular Basis of Mutualism and Antagonism in Epichloë spp. Mol. Plant-Microbe Interact. 2015, 28, 218–231. [Google Scholar]
- Johnson, L.J.; Johnson, R.D.; Schardl, C.L.; Panaccione, D.G. Identification of differentially expressed genes in the mutualistic association of tall fescue with Neotyphodium coenophialum. Physiol. Mol. Plant Pathol. 2003, 63, 305–317. [Google Scholar] [CrossRef]
- Khan, A.; Bassett, S.; Voisey, C.; Gaborit, C.; Johnson, L.; Christensen, M.; McCulloch, A.; Bryan, G.; Johnson, R. Gene expression profiling of the endophytic fungus Neotyphodium lolii in association with its host plant perennial ryegrass. Australas. Plant Pathol. 2010, 39, 467–476. [Google Scholar] [CrossRef]
- Dupont, P.-Y.; Eaton, C.J.; Wargent, J.J.; Fechtner, S.; Solomon, P.; Schmid, J.; Day, R.C.; Scott, B.; Cox, M.P. Fungal endophyte infection of ryegrass reprograms host metabolism and alters development. New Phytol. 2015, 208, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Eaton, C.J.; Cox, M.P.; Ambrose, B.; Becker, M.; Hesse, U.; Schardl, C.L.; Scott, B. Disruption of signaling in a fungal-grass symbiosis leads to pathogenesis. Plant Physiol. 2010, 153, 1780–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, Ö.; Braus, G.H. Coordination of secondary metabolism and development in fungi: The velvet family of regulatory proteins. FEMS Microbiol. Rev. 2012, 36, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Y.L.; Gerke, J.; Park, H.-S.; Bayram, Ö.; Neumann, P.; Ni, M.; Dickmanns, A.; Kim, S.C.; Yu, J.-H.; Braus, G.H.; et al. The Velvet Family of Fungal Regulators Contains a DNA-Binding Domain Structurally Similar to NF-κB. PLoS Biol. 2013, 11, e1001750. [Google Scholar] [CrossRef] [Green Version]
- Hoff, B.; Kamerewerd, J.; Sigl, C.; Mitterbauer, R.; Zadra, I.; Kürnsteiner, H.; Kück, U. Two Components of a velvet-Like Complex Control Hyphal Morphogenesis, Conidiophore Development, and Penicillin Biosynthesis in Penicillium chrysogenum. Eukaryot. Cell 2010, 9, 1236–1250. [Google Scholar] [CrossRef] [Green Version]
- Cary, J.W.; Obrian, G.R.; Nielsen, D.M.; Nierman, W.; Harris-Coward, P.; Yu, J.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Calvo, A.M. Elucidation of veA-dependent genes associated with aflatoxin and sclerotial production in Aspergillus flavus by functional genomics. Appl. Microbiol. Biotechnol. 2007, 76, 1107–1118. [Google Scholar] [CrossRef]
- Wiemann, P.; Brown, D.W.; Kleigrewe, K.; Bok, J.W.; Keller, N.P.; Humpf, H.-U.; Tudzynski, B. FfVel1 and FfLae1, components of a velvet-like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol. Microbiol. 2010, 77, 972–994. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, S.; Lind, A.L.; Lin, H.-C.; Tang, Y.; Rokas, A.; Calvo, A.M. The Fumagillin Gene Cluster, an Example of Hundreds of Genes under veA Control in Aspergillus fumigatus. PLoS ONE 2013, 8, e77147. [Google Scholar] [CrossRef] [Green Version]
- Lind, A.L.; Wisecaver, J.H.; Smith, T.D.; Feng, X.; Calvo, A.M.; Rokas, A. Examining the Evolution of the Regulatory Circuit Controlling Secondary Metabolism and Development in the Fungal Genus Aspergillus. PLoS Genet. 2015, 11, e1005096. [Google Scholar] [CrossRef] [PubMed]
- Cary, J.W.; Han, Z.; Yin, Y.; Lohmar, J.M.; Shantappa, S.; Harris-Coward, P.Y.; Mack, B.; Ehrlich, K.C.; Wei, Q.; Arroyo-Manzanares, N.; et al. Transcriptome Analysis of Aspergillus flavus Reveals veA-Dependent Regulation of Secondary Metabolite Gene Clusters, Including the Novel Aflavarin Cluster. Eukaryot. Cell 2015, 14, 983–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Liu, X.; Yin, Y.; Ma, Z. Involvement of a Velvet Protein FgVeA in the Regulation of Asexual Development, Lipid and Secondary Metabolisms and Virulence in Fusarium graminearum. PLoS ONE 2011, 6, e28291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, J.; Pradier, J.-M.; Simon, A.; Traeger, S.; Moraga, J.; Collado, I.G.; Viaud, M.; Tudzynski, B. Natural Variation in the VELVET Gene bcvel1 Affects Virulence and Light-Dependent Differentiation in Botrytis cinerea. PLoS ONE 2012, 7, e47840. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, J.; Simon, A.; Cohrs, K.C.; Traeger, S.; Porquier, A.; Dalmais, B.; Viaud, M.; Tudzynski, B. The VELVET complex in the gray mold fungus Botrytis cinerea: Impact of BcLAE1 on differentiation, secondary metabolism and virulence. Mol. Plant-Microbe Interact. 2015, 28, 659–674. [Google Scholar] [CrossRef] [Green Version]
- Bayram, O.; Krappmann, S.; Ni, M.; Bok, J.W.; Helmstaedt, K.; Valerius, O.; Braus-Stromeyer, S.A.; Kwon, N.-J.; Keller, N.P.; Yu, J.-H.; et al. VelB/VeA/LaeA Complex Coordinates Light Signal with Fungal Development and Secondary Metabolism. Science 2008, 320, 1504–1506. [Google Scholar] [CrossRef]
- Rahnama, M.; MacLean, P.; Fleetwood, D.; Johnson, R. Comparative transcriptomics analysis of compatible wild type and incompatible ΔlaeA mutant strains of Epichloë festucae in association with perennial ryegrass. Data Brief 2019, 24, 103843. [Google Scholar] [CrossRef]
- Unkles, S.E.; Hawker, K.L.; Grieve, C.; Campbell, E.I.; Montague, P.; Kinghorn, J.R. crnA encodes a nitrate transporter in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 1991, 88, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Mitsuzawa, H. Ammonium transporter genes in the fission yeast Schizosaccharomyces pombe: Role in ammonium uptake and a morphological transition. Genes Cells 2006, 11, 1183–1195. [Google Scholar] [CrossRef]
- Solomon, P.S.; Thomas, S.W.; Spanu, P.; Oliver, R.P. The utilisation of di/tripeptides by Stagonospora nodorum is dispensable for wheat infection. Physiol. Mol. Plant Pathol. 2003, 63, 191–199. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant Cell Wall—Degrading Enzymes and Their Secretion in Plant-Pathogenic Fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, K.-T.; Jeon, J.; Lee, Y.-H. Fungal plant cell wall-degrading enzyme database: A platform for comparative and evolutionary genomics in fungi and Oomycetes. BMC Genom. 2013, 14 (Suppl. 5), S7. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Giesbert, S.; Lepping, H.-B.; Tenberge, K.B.; Tudzynski, P. The Xylanolytic System of Claviceps purpurea: Cytological Evidence for Secretion of Xylanases in Infected Rye Tissue and Molecular Characterization of Two Xylanase Genes. Phytopathology 1998, 88, 1020–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christgau, S.; Kofod, L.V.; Halkier, T.; Andersen, L.N.; Hockauf, M.; Dörreich, K.; Dalbøge, H.; Kauppinen, S. Pectin methyl esterase from Aspergillus aculeatus: Expression cloning in yeast and characterization of the recombinant enzyme. Biochem. J. 1996, 319 Pt 3, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Leger, R.J.S.; Charnley, A.K.; Cooper, R.M. Characterization of cuticle-degrading proteases produced by the entomopathogen Metarhizium anisopliae. Arch. Biochem. Biophys. 1987, 253, 221–232. [Google Scholar] [CrossRef]
- Monaghan, J.; Zipfel, C. Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Segonzac, C.; Zipfel, C. Activation of plant pattern-recognition receptors by bacteria. Curr. Opin. Microbiol. 2011, 14, 54–61. [Google Scholar] [CrossRef]
- Bowman, S.M.; Free, S.J. The structure and synthesis of the fungal cell wall. BioEssays 2006, 28, 799–808. [Google Scholar] [CrossRef]
- Jaques, A.K.; Fukamizo, T.; Hall, D.; Barton, R.C.; Escott, G.M.; Parkinson, T.; Hitchcock, C.A.; Adams, D.J. Disruption of the gene encoding the ChiB1 chitinase of Aspergillus fumigatus and characterization of a recombinant gene product. Microbiology 2003, 149, 2931–2939. [Google Scholar] [CrossRef] [Green Version]
- Yarden, O.; Yanofsky, C. Chitin synthase 1 plays a major role in cell wall biogenesis in Neurospora crassa. Genes Dev. 1991, 5, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Horiuchi, H.; Ohta, A.; Takagi, M. Isolation and Characterization of EPD1, an Essential Gene for Pseudohyphal Growth of a Dimorphic Yeast, Candida maltosa. J. Bacteriol. 1998, 180, 2079–2086. [Google Scholar] [PubMed]
- Koch, M.R.; Pillus, L. The glucanosyltransferase Gas1 functions in transcriptional silencing. Proc. Natl. Acad. Sci. USA 2009, 106, 11224–11229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, Y.; Oda, M. Molecular characterization of endo-1,3-β-glucanase from Cellulosimicrobium cellulans: Effects of carbohydrate-binding module on enzymatic function and stability. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2011, 1814, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Shimoi, H.; Kitagaki, H.; Ohmori, H.; Iimura, Y.; Ito, K. Sed1p Is a Major Cell Wall Protein of Saccharomyces cerevisiae in the Stationary Phase and Is Involved in Lytic Enzyme Resistance. J. Bacteriol. 1998, 180, 3381–3387. [Google Scholar]
- Celerin, M.; Ray, J.M.; Schisler, N.J.; Day, A.W.; Stetler-Stevenson, W.G.; Laudenbach, D.E. Fungal fimbriae are composed of collagen. EMBO J. 1996, 15, 4445–4453. [Google Scholar] [CrossRef]
- Lai, J.; Koh, C.H.; Tjota, M.; Pieuchot, L.; Raman, V.; Chandrababu, K.B.; Yang, D.; Wong, L.; Jedd, G. Intrinsically disordered proteins aggregate at fungal cell-to-cell channels and regulate intercellular connectivity. Proc. Natl. Acad. Sci. USA 2012, 109, 15781–15786. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, D.J.; Scott, B.; Lane, G.A.; Tanaka, A.; Johnson, R.D. A Complex Ergovaline Gene Cluster in Epichloë Endophytes of Grasses. Appl. Environ. Microbiol. 2007, 73, 2571–2579. [Google Scholar] [CrossRef] [Green Version]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Rahnama, M.; Fields, K.A. Transformation of Chlamydia: Current approaches and impact on our understanding of chlamydial infection biology. Microbes Infect. 2018, 20, 445–450. [Google Scholar] [CrossRef]
- Hassing, B.; Winter, D.; Becker, Y.; Mesarich, C.H.; Eaton, C.J.; Scott, B. Analysis of Epichloë festucae small secreted proteins in the interaction with Lolium perenne. PLoS ONE 2019, 14, e0209463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Xiao, G.; Zheng, P.; Shang, Y.; Su, Y.; Zhang, X.; Liu, X.; Zhan, S.; Leger, R.J.S.; Wang, C. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 16796–16801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Liu, Z.; Lin, R.; Li, E.; Mao, Z.; Ling, J.; Yang, Y.; Yin, W.-B.; Xie, B. Biosynthesis of Antibiotic Leucinostatins in Bio-control Fungus Purpureocillium lilacinum and Their Inhibition on Phytophthora Revealed by Genome Mining. PLoS Pathog. 2016, 12, e1005685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, A.M.; Cary, J.W. Association of fungal secondary metabolism and sclerotial biology. Front. Microbiol. 2015, 6, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butchko, R.A.; Brown, D.W.; Busman, M.; Tudzynski, B.; Wiemann, P. Lae1 regulates expression of multiple secondary metabolite gene clusters in Fusarium verticillioides. Fungal Genet. Biol. 2012, 49, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, R.M.; Fedorova, N.D.; Bok, J.W.; Cramer, R.A.; Wortman, J.R.; Kim, H.S.; Nierman, W.C.; Keller, N.P. Transcriptional Regulation of Chemical Diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 2007, 3, e50. [Google Scholar] [CrossRef] [Green Version]
- Seiboth, B.; Karimi, R.A.; Phatale, P.A.; Linke, R.; Hartl, L.; Sauer, D.G.; Smith, K.M.; Baker, S.E.; Freitag, M.; Kubicek, C.P. The putative protein methyltransferase LAE1 controls cellulase gene expression in Trichoderma reesei. Mol. Microbiol. 2012, 84, 1150–1164. [Google Scholar] [CrossRef]
- Krappmann, S.; Braus, G.H. Nitrogen metabolism of Aspergillus and its role in pathogenicity. Med. Mycol. 2005, 43 (Suppl. 1), S31–S40. [Google Scholar] [CrossRef] [Green Version]
- Pellier, A.-L.; Laugé, R.; Veneault-Fourrey, C.; Langin, T. CLNR1, the AREA/NIT2-like global nitrogen regulator of the plant fungal pathogen Colletotrichum lindemuthianum is required for the infection cycle. Mol. Microbiol. 2003, 48, 639–655. [Google Scholar] [CrossRef]
- Rahnama, M.; Forester, N.; Ariyawansa, K.G.S.U.; Voisey, C.R.; Johnson, L.J.; Johnson, R.D.; Fleetwood, D.J. Efficient targeted mutagenesis in Epichloë festucae using a split marker system. J. Microbiol. Methods 2017, 134, 62–65. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hülsen, T.; De Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | In Culture (IC) | In Seedling (S) | In Planta (IP) |
|---|---|---|---|
| Host | PDA medium covered by cellophane | PRG seedlings | Mature infected PRG |
| Light condition | D | 8 h L & 16 h D | 8 h L & 16 h D |
| Time | 14 d | 14 dpi | 3 months post inoculation |
| E. festucae strains Comparisons |  |  |  |
| Fold Change | |||||||
|---|---|---|---|---|---|---|---|
| Model | Predicted Function | IC ∆laeA-WT | S ∆laeA-WT | IC ∆velA-WT | S ∆velA-WT | IP ∆velA-WT | Presence in Core Set |
| EfM3.003420 | Phospholipid transporter | −1.1 | −1.2 | −1.1 | −1.1 | 2.5 | No |
| EfM3.007320 | Copper transporter | 1.4 | 1.6 | 1.2 | 2.8 | 1.9 | No |
| EfM3.009680 | ABC transporter | −1.9 | −1.8 | −1.9 | −1.7 | 1.2 | No |
| EfM3.009730 | ABC transporter | −2.0 | -2.2 | −1.7 | −1.8 | −2.1 | No |
| EfM3.012390 | Nitrate transporter | 1.6 | 1.0 | 1.3 | 1.0 | 4.9 | No |
| EfM3.012760 | Malic acid transporter | 1.5 | −1.7 | −1.0 | −3.0 | −1.4 | Yes |
| EfM3.014790 | ABC transporter | 1.2 | −1.8 | −2.1 | −1.9 | −1.4 | No |
| EfM3.018210 | Proline-specific permease | −1.1 | 1.1 | −1.0 | 1.2 | −2.2 | No |
| EfM3.020140 | Sulphate transporter | 1.1 | 1.4 | 1.2 | 2.5 | −1.7 | No |
| EfM3.025050 | Amino acid permease | 1.4 | 1.3 | 1.1 | 2.2 | 1.1 | No |
| EfM3.025350 | Transporter | −1.3 | −1.3 | −1.7 | 1.2 | −1.1 | No |
| EfM3.027520 | ABC transporter | 1.1 | 1.7 | 1.2 | 3.1 | 2.8 | No |
| EfM3.027540 | ABC transporter | 1.3 | 1.4 | 1.4 | 2.5 | 2.8 | No |
| EfM3.027570 | Peptide transporter | 2.6 | 3.1 | 3.0 | 9.9 | 12.5 | Yes |
| EfM3.032550 | ABC transporter | 1.2 | 1.3 | 1.1 | −1.2 | 1.2 | No |
| EfM3.035410 | Carboxylic acid transporter | 1.4 | 2.0 | −1.1 | −1.0 | 5.1 | Yes |
| EfM3.039020 | Ammonium transporter | 1.0 | 2.0 | 1.0 | 3.0 | 4.5 | No |
| EfM3.040210 | Phospholipid-translocating ATPase | −1.2 | −1.1 | −1.1 | −1.1 | 2.4 | No |
| EfM3.045520 | Hydrogen ion transmembrane transporter | 3.4 | −2.4 | 8.6 | −8.4 | −4.0 | No |
| EfM3.047210 | Purine permease | −1.1 | 1.2 | −1.1 | 1.3 | 2.1 | No |
| EfM3.055090 | ABC multidrug transporter | 1.1 | −1.2 | −1.1 | −2.7 | 1.3 | No |
| EfM3.056220 | ABC multidrug transporter | 1.2 | −4.8 | −1.1 | −4.2 | −3.4 | Yes |
| EfM3.058970 | Na+/H+ antiporter | 1.0 | −1.1 | −1.1 | −1.1 | −2.1 | No |
| EfM3.066900 | Xanthine/uracil permease | 1.1 | −1.6 | 1.4 | −2.9 | -6.1 | Yes |
| EfM3.074200 | Transmembrane transporter | 2.0 | −1.3 | 2.8 | −2.6 | −1.6 | No |
| EfM3.075680 | Transmembrane transporter | −1.8 | −2.6 | −2.1 | −2.4 | −2.4 | No |
| EfM3.000930 | Zinc ion transporter | 1.1 | −2.8 | −1.1 | −1.4 | 1.1 | No |
| EfM3.005950 | Amino acid permease | 1.4 | 2.4 | −1.0 | 1.5 | 1.4 | No |
| EfM3.029800 | ABC multidrug transporter | 2.4 | 1.1 | 1.1 | −1.5 | −1.1 | No |
| EfM3.064820 | Sugar transporter | −1.7 | −2.4 | −1.2 | 1.2 | −1.2 | No |
| EfM3.077030 | Inositol transporter | 1.3 | 2.2 | 1.2 | 1.4 | 1.1 | No |
| EfM3.080140 | Calcium transporter | −1.1 | −2.4 | −1.0 | −1.6 | 1.8 | No |
| EfM3.158840 | Metal ion transporter | 1.1 | 2.0 | 1.0 | 2.0 | 1.1 | No |
| Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|
| Model | CAZyme Class | Predicted Function | Target Cell Wall Component | IC ∆laeA-WT | S ∆laeA-WT | IC ∆velA-WT | S ∆velA-WT | IP ∆velA-WT |
| EfM3.049570 | GH13 | Alpha-glucosidase | Cellulose | 1.0 | −2.1 | −1.2 | −1.5 | −1.2 |
| EfM3.053990 | GH5 | Endoglucanase | Cellulose | 1.5 | 2.1 | −1.1 | 1.1 | 1.3 |
| EfM3.005420 | N/A | Exo-1,4-beta-xylosidase | Hemicellulose | 1.1 | 1.9 | −1.0 | 2.3 | 1.5 |
| EfM3.040190 | GH10 | Endo-1,4-beta-xylanase | Hemicellulose | 1.7 | 3.3 | 1.1 | 3.9 | 83.5 |
| EfM3.037040 | GH62 | Glycosyl hydrolase | Hemicellulose | 1.1 | 4.3 | 1.0 | 4.3 | 69.6 |
| EfM3.008730 | CE8 | Pectin methylesterase | Pectin | −1.3 | 1.7 | 1.0 | 2.0 | 1.4 |
| EfM3.008610 | CE5 | Cutinase | Cutin | 2.5 | 2.9 | 1.2 | −2.0 | 2.9 |
| EfM3.005300 | N/A | Cuticle-degrading protease | Cuticle | 1.0 | −2.8 | 1.0 | −5.1 | −4.1 |
| Fold Change | |||||||
|---|---|---|---|---|---|---|---|
| Model | CAZyme Class | Predicted Function | IC ∆laeA-WT | S ∆laeA-WT | IC ∆velA-WT | S ∆velA-WT | IP ∆velA-WT |
| EfM3.000810 | CBM18 | Chitinase | 1.0 | −3.0 | 1.0 | −3.3 | −3.1 |
| EfM3.024310 | GH18 | Endochitinase B1 | 1.4 | 1.6 | −1.1 | 1.9 | 2.1 |
| EfM3.049120 | GT2 | Chitin synthase | −1.3 | −1.4 | 2.0 | 1.3 | −1.4 |
| EfM3.056450 | CBM43 | Glucanosyl transferase | −1.2 | −1.2 | −1.0 | −1.2 | 2.1 |
| EfM3.056810 | GH64 | Glucan endo-1,3-beta-glucosidase | 1.1 | 1.1 | −1.1 | 1.0 | −2.2 |
| EfM3.078790 | N/A | Cell wall protein SED1 | 1.1 | −3.4 | 1.1 | −2.5 | −1.8 |
| EfM3.054000 | N/A | Related to cell wall glycoprotein | −1.7 | 1.1 | 1.1 | 2.3 | 1.9 |
| EfM3.034340 | N/A | Cell wall glycoprotein | −1.2 | −3.4 | 1.4 | −1.9 | −3.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahnama, M.; Maclean, P.; Fleetwood, D.J.; Johnson, R.D. VelA and LaeA are Key Regulators of Epichloë festucae Transcriptomic Response during Symbiosis with Perennial Ryegrass. Microorganisms 2020, 8, 33. https://doi.org/10.3390/microorganisms8010033
Rahnama M, Maclean P, Fleetwood DJ, Johnson RD. VelA and LaeA are Key Regulators of Epichloë festucae Transcriptomic Response during Symbiosis with Perennial Ryegrass. Microorganisms. 2020; 8(1):33. https://doi.org/10.3390/microorganisms8010033
Chicago/Turabian StyleRahnama, Mostafa, Paul Maclean, Damien J. Fleetwood, and Richard D. Johnson. 2020. "VelA and LaeA are Key Regulators of Epichloë festucae Transcriptomic Response during Symbiosis with Perennial Ryegrass" Microorganisms 8, no. 1: 33. https://doi.org/10.3390/microorganisms8010033