Rapid Changes in Microbial Community Structures along a Meandering River

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
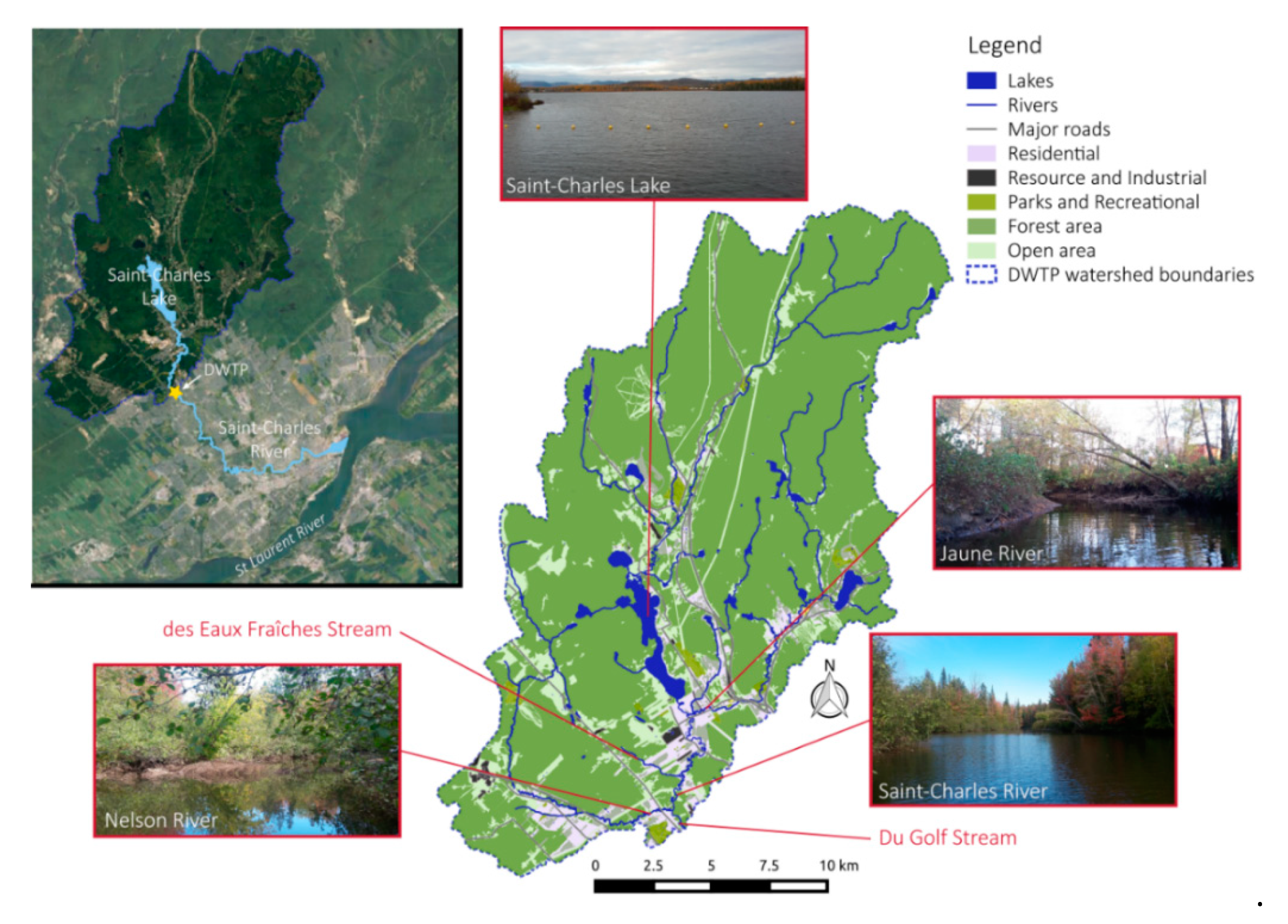
2.1. Study Area
2.2. Sampling Sites and Methods
2.3. DNA Extraction, DNA Amplification, and Sequencing
2.4. Sequencing Analyses
2.5. Quantitative Real-Time PCR
2.6. Physicochemical Parameters
2.7. Statistical Analyses
3. Results
3.1. Environmental Parameters
3.2. Microbial Community Structures
3.3. Presence/Absence of OTUs
3.4. Correlations between Bacterial OTUs, Eukaryotic OTUs, and Environmental Parameters
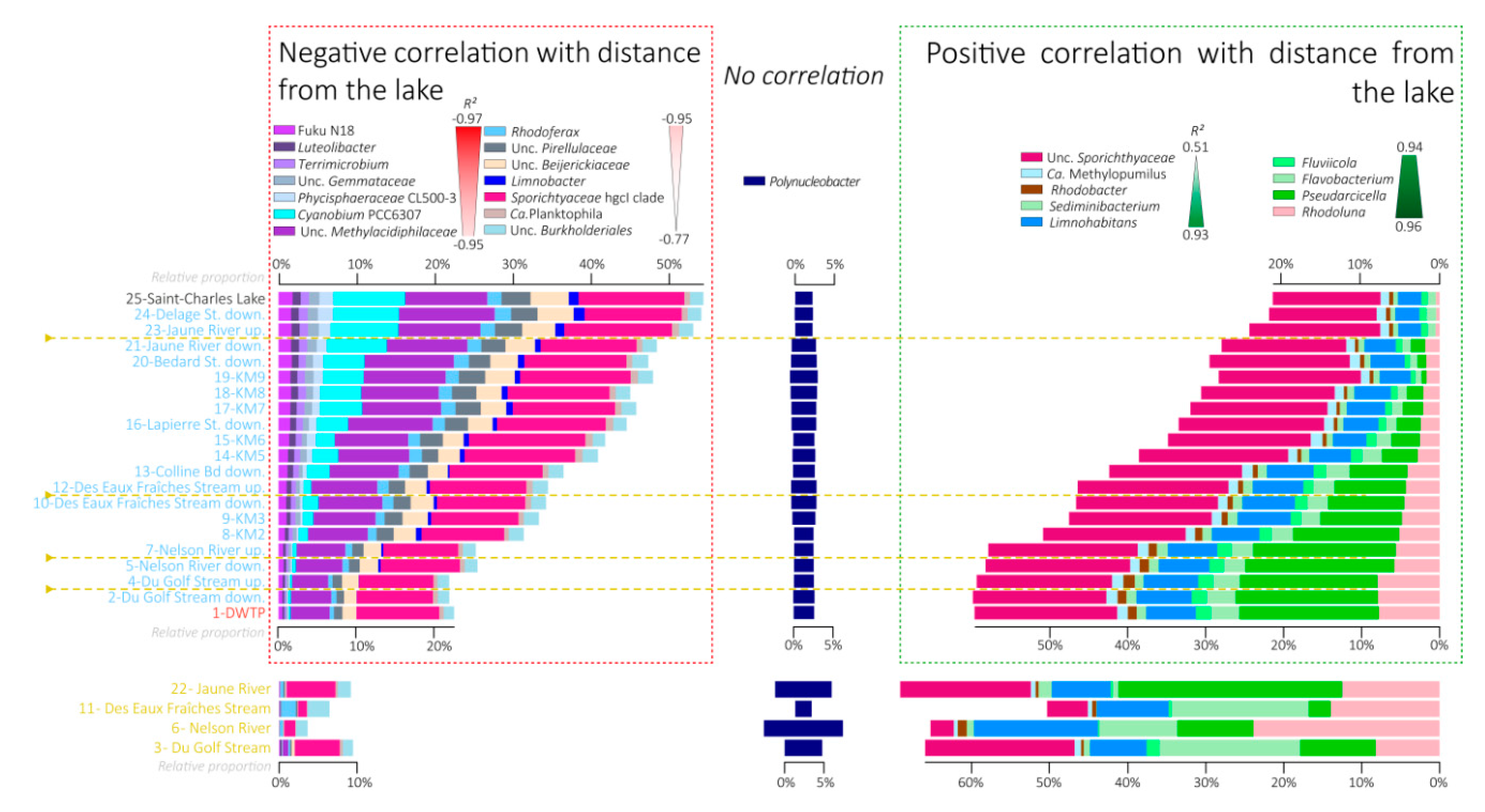
3.5. Variations in the Relative Proportions of the Main Bacterial Taxa
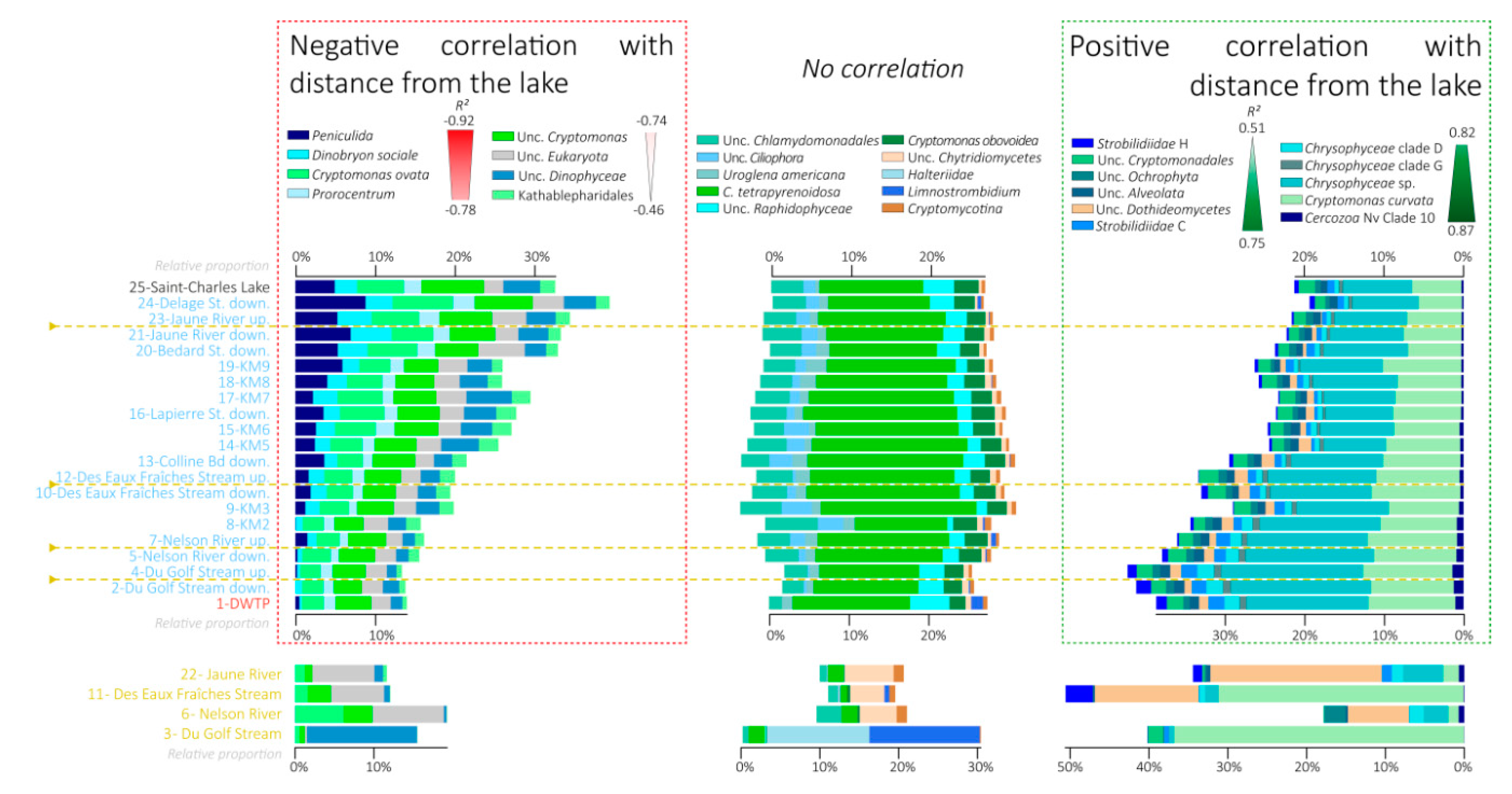
3.6. Variations in the Relative Proportions of the Main Eukaryotic Taxa
3.7. Quantitative Real-Time PCR
4. Discussion
4.1. Progressive Changes of the Global Microbial Community along the River Course
4.2. Contrasted Dynamics among Microbial Communities
4.3. Fall of the Lake Microbial Communities
4.4. Rise of the Riverine Microbial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stockner, J.G. Phototrophic picoplankton: An overview from marine and freshwater ecosystems. Limnol. Oceanogr. 1988, 33, 765–775. [Google Scholar] [CrossRef]
- Caron, D.A. Inorganic nutrients, bacteria, and the microbial loop. Microb. Ecol. 1994, 28, 295–298. [Google Scholar] [CrossRef]
- Finlay, B.; Esteban, G. Freshwater protozoa: Biodiversity and ecological function. Biodivers. Conserv. 1998, 7, 1163–1186. [Google Scholar] [CrossRef]
- Arndt, H.; Dietrich, D.; Auer, B.; Cleven, E.-J.; Gräfenhan, T.; Weitere, M.; Mylnikov, A.P. Functional diversity of heterotrophic flagellates in aquatic ecosystems. In The Flagellates: Unity, Diversity and Evolution; Taylor & Francis: London, UK, 2000; pp. 240–268. [Google Scholar]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, E.; Sherr, B. Role of microbes in pelagic food webs: A revised concept. Limnol. Oceanogr. 1988, 33, 1225–1227. [Google Scholar] [CrossRef]
- Sanders, R.W.; Wickham, S.A. Planktonic protozoa and metazoa: Predation, food quality and population control. Mar. Microb. Food Webs 1993, 7, 197–223. [Google Scholar]
- Crump, B.C.; Armbrust, E.V.; Baross, J.A. Phylogenetic Analysis of Particle-Attached and Free-Living Bacterial Communities in the Columbia River, Its Estuary, and the Adjacent Coastal Ocean. Appl. Environ. Microbiol. 1999, 65, 3192–3204. [Google Scholar] [CrossRef] [Green Version]
- Zwart, G.; Crump, B.C.; Agterveld, M.P.K.; Hagen, F.; Han, S.-K. Typical freshwater bacteria: An analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 2002, 28, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Cruaud, P.; Vigneron, A.; Fradette, M.-S.; Dorea, C.C.; Culley, A.I.; Rodriguez, M.J.; Charette, S.J. Annual bacterial community cycle in a seasonally ice-covered river reflects environmental and climatic conditions. Limnol. Oceanogr. 2019. [Google Scholar] [CrossRef]
- Richards, T.A.; Vepritskiy, A.A.; Gouliamova, D.E.; Nierzwicki-Bauer, S.A. The molecular diversity of freshwater picoeukaryotes from an oligotrophic lake reveals diverse, distinctive and globally dispersed lineages. Environ. Microbiol. 2005, 7, 1413–1425. [Google Scholar] [CrossRef]
- Šlapeta, J.; Moreira, D.; López-García, P. The extent of protist diversity: Insights from molecular ecology of freshwater eukaryotes. Proc. R. Soc. Lond. B Biol. Sci. 2005, 272, 2073–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; López-García, P.; Deschamps, P.; Moreira, D.; Restoux, G.; Bertolino, P.; Jardillier, L. Marked seasonality and high spatial variability of protist communities in shallow freshwater systems. ISME J. 2015, 9, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruaud, P.; Vigneron, A.; Fradette, M.-S.; Dorea, C.C.; Culley, A.I.; Rodriguez, M.J.; Charette, S.J. Annual Protist Community Dynamics in a Freshwater Ecosystem Undergoing Contrasted Climatic Conditions: The Saint-Charles River (Canada). Front. Microbiol. 2019, 10, 2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savio, D.; Sinclair, L.; Ijaz, U.Z.; Parajka, J.; Reischer, G.H.; Stadler, P.; Blaschke, A.P.; Blöschl, G.; Mach, R.L.; Kirschner, A.K.T.; et al. Bacterial diversity along a 2600 km river continuum. Environ. Microbiol. 2015, 17, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Ensign, S.H.; Doyle, M.W. Nutrient spiraling in streams and river networks. J. Geophys. Res. Biogeosci. 2006, 111. [Google Scholar] [CrossRef]
- Cole, J.J.; Prairie, Y.T.; Caraco, N.F.; McDowell, W.H.; Tranvik, L.J.; Striegl, R.G.; Duarte, C.M.; Kortelainen, P.; Downing, J.A.; Middelburg, J.J.; et al. Plumbing the Global Carbon Cycle: Integrating Inland Waters into the Terrestrial Carbon Budget. Ecosystems 2007, 10, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Withers, P.J.A.; Jarvie, H.P. Delivery and cycling of phosphorus in rivers: A review. Sci. Total Environ. 2008, 400, 379–395. [Google Scholar] [CrossRef]
- Battin, T.J.; Luyssaert, S.; Kaplan, L.A.; Aufdenkampe, A.K.; Richter, A.; Tranvik, L.J. The boundless carbon cycle. Nature 2009, 2, 598–600. [Google Scholar] [CrossRef]
- Findlay, S.; Sinsabaugh, R.L. Unravelling the sources and bioavailability of dissolved organic matter in lotic aquatic ecosystems. Mar. Freshw. Res. 1999, 50, 781–790. [Google Scholar] [CrossRef]
- Caron, D.A.; Worden, A.Z.; Countway, P.D.; Demir, E.; Heidelberg, K.B. Protists are microbes too: A perspective. ISME J. 2009, 3, 4–12. [Google Scholar] [CrossRef]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Vannote, R.L.; Minshall, G.W.; Cummins, K.W.; Sedell, J.R.; Cushing, C.E. The River Continuum Concept. Can. J. Fish. Aquat. Sci. 1980, 37, 130–137. [Google Scholar] [CrossRef]
- Twiss, M.R.; Ulrich, C.; Kring, S.A.; Harold, J.; Williams, M.R. Plankton dynamics along a 180 km reach of the Saint Lawrence River from its headwaters in Lake Ontario. Hydrobiologia 2010, 647, 7–20. [Google Scholar] [CrossRef]
- Sekiguchi, H.; Watanabe, M.; Nakahara, T.; Xu, B.; Uchiyama, H. Succession of Bacterial Community Structure along the Changjiang River Determined by Denaturing Gradient Gel Electrophoresis and Clone Library Analysis. Appl. Environ. Microbiol. 2002, 68, 5142–5150. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.; Hein, T.; Kavka, G.; Mach, R.L.; Farnleitner, A.H. Longitudinal Changes in the Bacterial Community Composition of the Danube River: A Whole-River Approach. Appl. Environ. Microbiol. 2007, 73, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, D.S.; Gweon, H.S.; Bowes, M.J.; Newbold, L.K.; Field, D.; Bailey, M.J.; Griffiths, R.I. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 2015, 9, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, J. Origin and succession of phytoplankton in a river-lake system (Spree, Germany). In Phytoplankton in Turbid Environments: Rivers and Shallow Lakes, Proceedings of the 9th Workshop of the International Association of Phytoplankton Taxonomy and Ecology (IAP), Mont Rigi, Belgium, 10–18 July 1993; Descy, J.-P., Reynolds, C.S., Padisák, J., Eds.; Developments in Hydrobiology; Springer: Dordrecht, The Netherlands, 1994; pp. 73–83. ISBN 978-94-017-2670-2. [Google Scholar]
- Whitehead, P.G.; Crossman, J.; Balana, B.B.; Futter, M.N.; Comber, S.; Jin, L.; Skuras, D.; Wade, A.J.; Bowes, M.J.; Read, D.S. A cost-effectiveness analysis of water security and water quality: Impacts of climate and land-use change on the River Thames system. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2013, 371, 20120413. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, C.; Lewis, F.; Huet-Alegre, E.; Ksouri, Y.; Leclerc, M.-C.; Viens, D. Portrait du bassin de la rivière Saint-Charles-2ème édition-216 p. Cons. Bassin Rivière St.-Charles 2009, 9, 217–340. [Google Scholar]
- Gerardin, V.; Lachance, Y. Versune Gestion Intégrée des Bassins Versants. In Atlas du Cadre Écologique de Référence du Bassin Versant de la Rivière Saint-Charles, Québec, Canada, Min; de l’Environnement et de la Faune du Québec-Min, de l’Environnement du Canada: Outaouais, Québec, Canada, 1997; 58p. [Google Scholar]
- Boulé, V.; Vallières, C.; Laflamme, V.; Bouchard-Valentine, M.; Jobin, P.; Sauvageau, C.; Côté, P.; Pagé, N. État de la Situation du Bassin Versant de la Prise d’eau de la Rivière St-Charles-Rapport Final 2010; Roche: Laval, QC, Canada, 2010. [Google Scholar]
- Cruaud, P.; Vigneron, A.; Fradette, M.-S.; Charette, S.J.; Rodriguez, M.J.; Dorea, C.C.; Culley, A.I. Open the SterivexTM casing: An easy and effective way to improve DNA extraction yields. Limnol. Oceanogr. Methods 2017, 15, 1015–1020. [Google Scholar] [CrossRef]
- Cruaud, P.; Rasplus, J.-Y.; Rodriguez, L.J.; Cruaud, A. High-throughput sequencing of multiple amplicons for barcoding and integrative taxonomy. Sci. Rep. 2017, 7, 41948. [Google Scholar] [CrossRef] [Green Version]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, A.M.; Li, W.K.W.; Tremblay, J.-É.; Carmack, E.C.; Lovejoy, C. Arctic Ocean Microbial Community Structure before and after the 2007 Record Sea Ice Minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Application for High Throughput Sequence Data. 2012. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 October 2020).
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillou, L.; Bachar, D.; Audic, S.; Bass, D.; Berney, C.; Bittner, L.; Boutte, C.; Burgaud, G.; de Vargas, C.; Decelle, J.; et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013, 41, D597–D604. [Google Scholar] [CrossRef]
- Suzuki, M.T.; Taylor, L.T.; DeLong, E.F. Quantitative Analysis of Small-Subunit rRNA Genes in Mixed Microbial Populations via 5′-Nuclease Assays. Appl. Environ. Microbiol. 2000, 66, 4605–4614. [Google Scholar] [CrossRef] [Green Version]
- American Public Health Association; American Water Works Association; Water Environment Federation. Standard Methods for the Examination of Water and Wastewater, 22nd ed.; American Water Works Association: Denver, CO, USA, 2012. [Google Scholar]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M. Gplots: Various R programming tools for plotting data. R package version 2.17.0. Comput. Softw. 2015, 2, 1. [Google Scholar]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef] [PubMed]
- Leff, L.G.; Brown, B.J.; Lemke, M.J. Spatial and Temporal Changes in Bacterial Assemblages of the Cuyahoga River. Ohio J. Sci. 1999, 99, 44–48. [Google Scholar]
- Sukhodolov, A.; Thiele, M.; Bungartz, H. Turbulence structure in a river reach with sand bed. Water Resour. Res. 1998, 34, 1317–1334. [Google Scholar] [CrossRef]
- Lewis, M.R.; Horne, E.P.W.; Cullen, J.J.; Oakey, N.S.; Platt, T. Turbulent motions may control phytoplankton photosynthesis in the upper ocean. Nature 1984, 311, 49–50. [Google Scholar] [CrossRef]
- Battin, T.J.; Besemer, K.; Bengtsson, M.M.; Romani, A.M.; Packmann, A.I. The ecology and biogeochemistry of stream biofilms. Nat. Rev. Microbiol. 2016, 14, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Bouma-Gregson, K.; Olm, M.R.; Probst, A.J.; Anantharaman, K.; Power, M.E.; Banfield, J.F. Microbial diversity and metabolic potential in cyanotoxin producing cyanobacterial mats throughout a river network. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Hotaling, S.; Foley, M.E.; Zeglin, L.H.; Finn, D.S.; Tronstad, L.M.; Giersch, J.J.; Muhlfeld, C.C.; Weisrock, D.W. Microbial assemblages reflect environmental heterogeneity in alpine streams. Glob. Chang. Biol. 2019, 25, 2576–2590. [Google Scholar] [CrossRef]
- Brönmark, C.; Malmqvist, B. Spatial and temporal patterns of lake outlet benthos: With 6 figures and 1 table in the text. Int. Ver. Theor. Angew. Limnol. Verh. 1984, 22, 1986–1991. [Google Scholar] [CrossRef]
- Robinson, C.T.; Minshall, G.W. Longitudinal development of macroinvertebrate communities below oligotrophic lake outlets. Gt. Basin Nat. 1990, 50, 303–311. [Google Scholar]
- Welker, M.; Walz, N. Can mussels control the plankton in rivers?—A planktological approach applying a Lagrangian sampling strategy. Limnol. Oceanogr. 1998, 43, 753–762. [Google Scholar] [CrossRef]
- Cohen, R.R.H.; Dresler, P.V.; Phillips, E.J.P.; Cory, R.L. The effect of the Asiatic clam, Corbicula fluminea, on phytoplankton of the Potomac River, Maryland. Limnol. Oceanogr. 1984, 29, 170–180. [Google Scholar] [CrossRef]
- Richardson, J.S.; Mackay, R.J. Lake Outlets and the Distribution of Filter Feeders: An Assessment of Hypotheses. Oikos 1991, 62, 370–380. [Google Scholar] [CrossRef]
- Pusch, M.; Siefert, J.; Walz, N. Filtration and Respiration Rates of Two Unionid Species and Their Impact on the Water Quality of a Lowland River. In Ecology and Evolution of the Freshwater Mussels Unionoida; Bauer, G., Wächtler, K., Eds.; Ecological Studies; Springer: Berlin/Heidelberg, Germany, 2001; pp. 317–326. ISBN 978-3-642-56869-5. [Google Scholar]
- Flesch, J.C.; Spicer, P.T.; Pratsinis, S.E. Laminar and turbulent shear-induced flocculation of fractal aggregates. AIChE J. 1999, 45, 1114–1124. [Google Scholar] [CrossRef]
- Ruiz, J.; Macías, D.; Peters, F. Turbulence increases the average settling velocity of phytoplankton cells. Proc. Natl. Acad. Sci. USA 2004, 101, 17720–17724. [Google Scholar] [CrossRef] [Green Version]
- Verspagen, J.M.H.; Visser, P.M.; Huisman, J. Aggregation with clay causes sedimentation of the buoyant cyanobacteria Microcystis spp. Aquat. Microb. Ecol. 2006, 44, 165–174. [Google Scholar] [CrossRef]
- Jørgensen, C.B. Comparative Physiology of Suspension Feeding. Annu. Rev. Physiol. 1975, 37, 57–79. [Google Scholar] [CrossRef]
- Rubenstein, D.I.; Koehl, M.A.R. The Mechanisms of Filter Feeding: Some Theoretical Considerations. Am. Nat. 1977, 111, 981–994. [Google Scholar] [CrossRef]
- Bar-Or, Y.; Shilo, M. The role of cell-bound flocculants in coflocculation of benthic cyanobacteria with clay particles. FEMS Microbiol. Ecol. 1988, 4, 169–174. [Google Scholar] [CrossRef]
- Ibelings, B.W.; Bruin, A.D.; Kagami, M.; Rijkeboer, M.; Brehm, M.; Donk, E.V. Host Parasite Interactions Between Freshwater Phytoplankton and Chytrid Fungi (chytridiomycota). J. Phycol. 2004, 40, 437–453. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.; Brazel, D.M.; Swan, B.K.; Arnosti, C.; Chain, P.S.G.; Reitenga, K.G.; Xie, G.; Poulton, N.J.; Gomez, M.L.; Masland, D.E.D.; et al. Capturing Single Cell Genomes of Active Polysaccharide Degraders: An Unexpected Contribution of Verrucomicrobia. PLoS ONE 2012, 7, e35314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlemann, D.P.R.; Lundin, D.; Labrenz, M.; Jürgens, K.; Zheng, Z.; Aspeborg, H.; Andersson, A.F. Metagenomic De Novo Assembly of an Aquatic Representative of the Verrucomicrobial Class Spartobacteria. mBio 2013, 4, e00569-12. [Google Scholar] [CrossRef] [Green Version]
- Pascual, J.; García-López, M.; González, I.; Genilloud, O. Luteolibactergellanilyticus sp. nov., a gellan-gum-degrading bacterium of the phylum Verrucomicrobia isolated from miniaturized diffusion chambers. Int. J. Syst. Evol. Microbiol. 2017, 67, 3951–3959. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Ramachandran, A.; Khawasek, O.; Beisner, B.E.; Rautio, M.; Huot, Y.; Walsh, D.A. Microbial life under ice: Metagenome diversity and in situ activity of Verrucomicrobia in seasonally ice-covered lakes. Environ. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, A.; Schmidt, J.; Koll, U.; Hahn, M.W. Aquirufaantheringensis gen. nov., sp. nov. and Aquirufanivalisilvae sp. nov., representing a new genus of widespread freshwater bacteria. Int. J. Syst. Evol. Microbiol. 2019, 69, 2739–2749. [Google Scholar] [CrossRef]
- Zimmermann-Timm, H. Characteristics, Dynamics and Importance of Aggregates in Rivers–An Invited Review. Int. Rev. Hydrobiol. 2002, 87, 197–240. [Google Scholar] [CrossRef]
- Pusch, M.; Fiebig, D.; Brettar, I.; Eisenmann, H.; Ellis, B.K.; Kaplan, L.A.; Lock, M.A.; Naegeli, M.W.; Traunspurger, W. The role of micro-organisms in the ecological connectivity of running waters. Freshw. Biol. 1998, 40, 453–495. [Google Scholar] [CrossRef]
- Reynolds, C.S. Hydroecology of river plankton: The role of variability in channel flow. Hydrol. Process. 2000, 14, 3119–3132. [Google Scholar] [CrossRef]
- Šimek, K.; Horňák, K.; Jezbera, J.; Nedoma, J.; Vrba, J.; Straškrábová, V.; Macek, M.; Dolan, J.R.; Hahn, M.W. Maximum growth rates and possible life strategies of different bacterioplankton groups in relation to phosphorus availability in a freshwater reservoir. Environ. Microbiol. 2006, 8, 1613–1624. [Google Scholar] [CrossRef]
- Kasalický, V.; Jezbera, J.; Simek, K.; Hahn, M.W. Limnohabitansplanktonicus sp. nov. and Limnohabitansparvus sp. nov., planktonic betaproteobacteria isolated from a freshwater reservoir, and emended description of the genus Limnohabitans. Int. J. Syst. Evol. Microbiol. 2010, 60, 2710–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.R.; Brown, R.M. Cytophaga That Kills or Lyses Algae. Science 1969, 164, 1523–1524. [Google Scholar] [CrossRef] [PubMed]
- Daft, M.J.; McCord, S.B.; Stewart, W.D.P. Ecological studies on algal-lysing bacteria in fresh waters. Freshw. Biol. 1975, 5, 577–596. [Google Scholar] [CrossRef]
- Rashidan, K.K.; Bird, D.F. Role of predatory bacteria in the termination of a cyanobacterial bloom. Microb. Ecol. 2001, 41, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Šimek, K.; Kasalický, V.; Jezbera, J.; Jezberová, J.; Hejzlar, J.; Hahn, M.W. Broad Habitat Range of the Phylogenetically Narrow R-BT065 Cluster, Representing a Core Group of the Betaproteobacterial Genus Limnohabitans. Appl. Environ. Microbiol. 2010, 76, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Margalef, R. Ideas for a Synthetic Approach to the Ecology of Running Waters. Int. Rev. Gesamten Hydrobiol. Hydrogr. 1960, 45, 133–153. [Google Scholar] [CrossRef]
- Reckendorfer, W.; Keckeis, H.; Winkler, G.; Schiemer, F. Zooplankton abundance in the River Danube, Austria: The significance of inshore retention. Freshw. Biol. 1999, 41, 583–591. [Google Scholar] [CrossRef]
- Speirs, D.C.; Gurney, W.S.C. Population Persistence in Rivers and Estuaries. Ecology 2001, 82, 1219–1237. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruaud, P.; Vigneron, A.; Dorea, C.C.; Rodriguez, M.J.; Charette, S.J. Rapid Changes in Microbial Community Structures along a Meandering River. Microorganisms 2020, 8, 1631. https://doi.org/10.3390/microorganisms8111631
Cruaud P, Vigneron A, Dorea CC, Rodriguez MJ, Charette SJ. Rapid Changes in Microbial Community Structures along a Meandering River. Microorganisms. 2020; 8(11):1631. https://doi.org/10.3390/microorganisms8111631
Chicago/Turabian StyleCruaud, Perrine, Adrien Vigneron, Caetano C. Dorea, Manuel J. Rodriguez, and Steve J. Charette. 2020. "Rapid Changes in Microbial Community Structures along a Meandering River" Microorganisms 8, no. 11: 1631. https://doi.org/10.3390/microorganisms8111631
APA StyleCruaud, P., Vigneron, A., Dorea, C. C., Rodriguez, M. J., & Charette, S. J. (2020). Rapid Changes in Microbial Community Structures along a Meandering River. Microorganisms, 8(11), 1631. https://doi.org/10.3390/microorganisms8111631