A Genomic Survey of Signalling in the Myxococcaceae



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Genome Sequences
2.2. Identification of Regulatory Proteins
3. Results
3.1. Selection of Sets of Genomes
3.2. MyxococcacealGenomes Encode Similar Numbers and Types of Regulatory Proteins
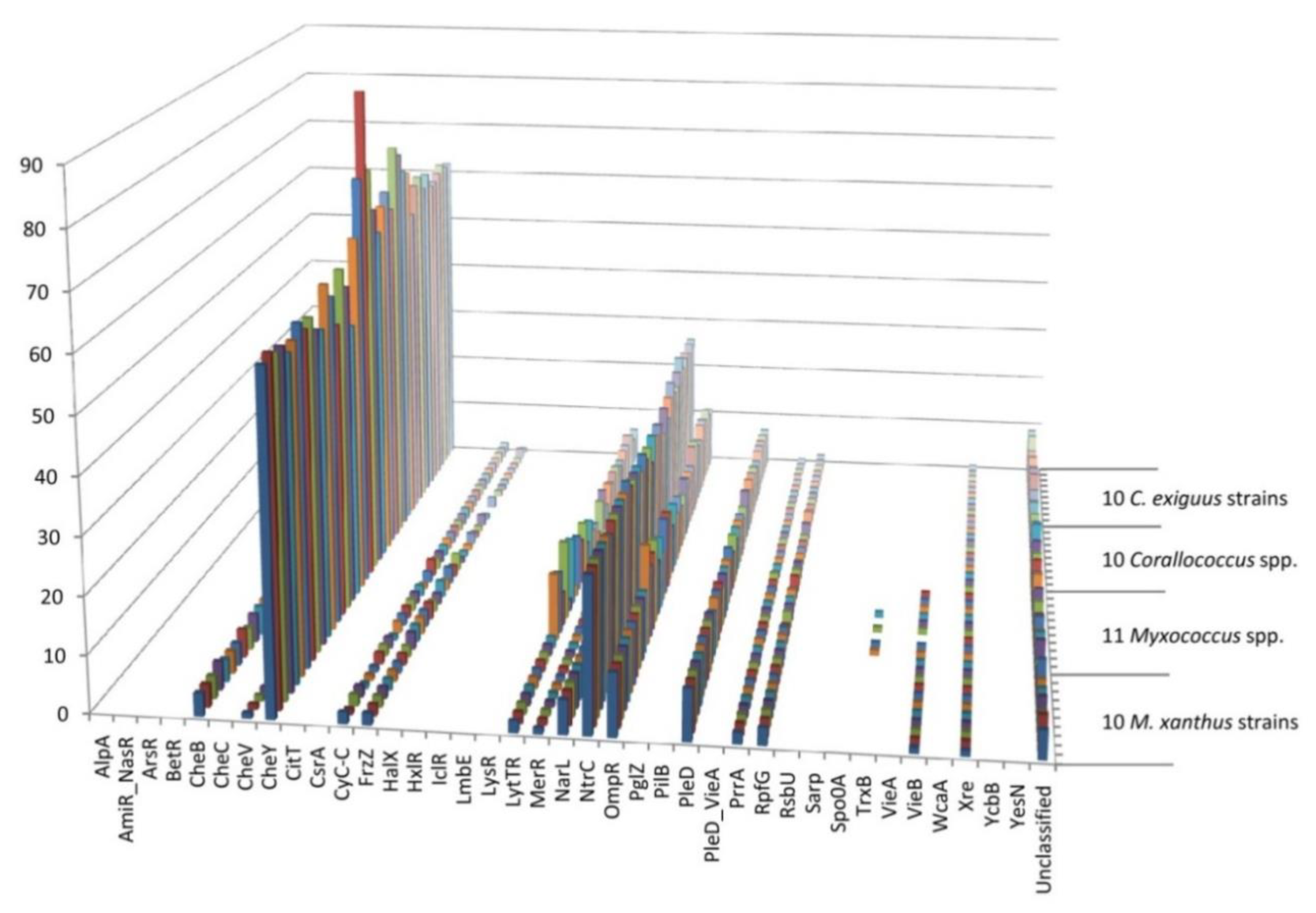
3.3. Different Families of Regulators Exhibit Distinct Patterns of Conservation
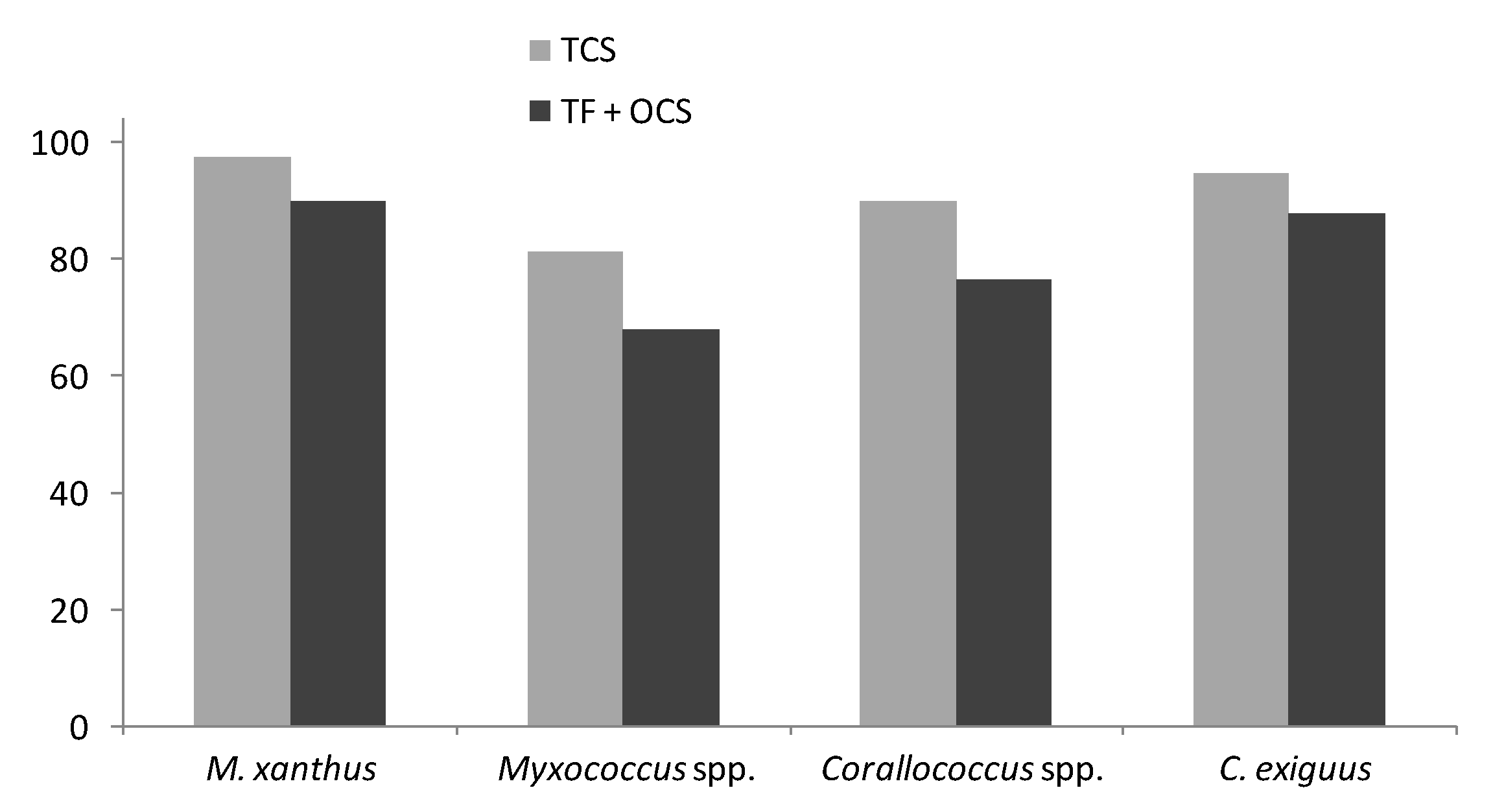
3.4. TCS Proteins are More Enriched in Myxobacterial Core Genomes than Other Regulatory Proteins
3.5. Conservation of Regulatory Proteins Involved in Key Myxococcaceal Behaviours
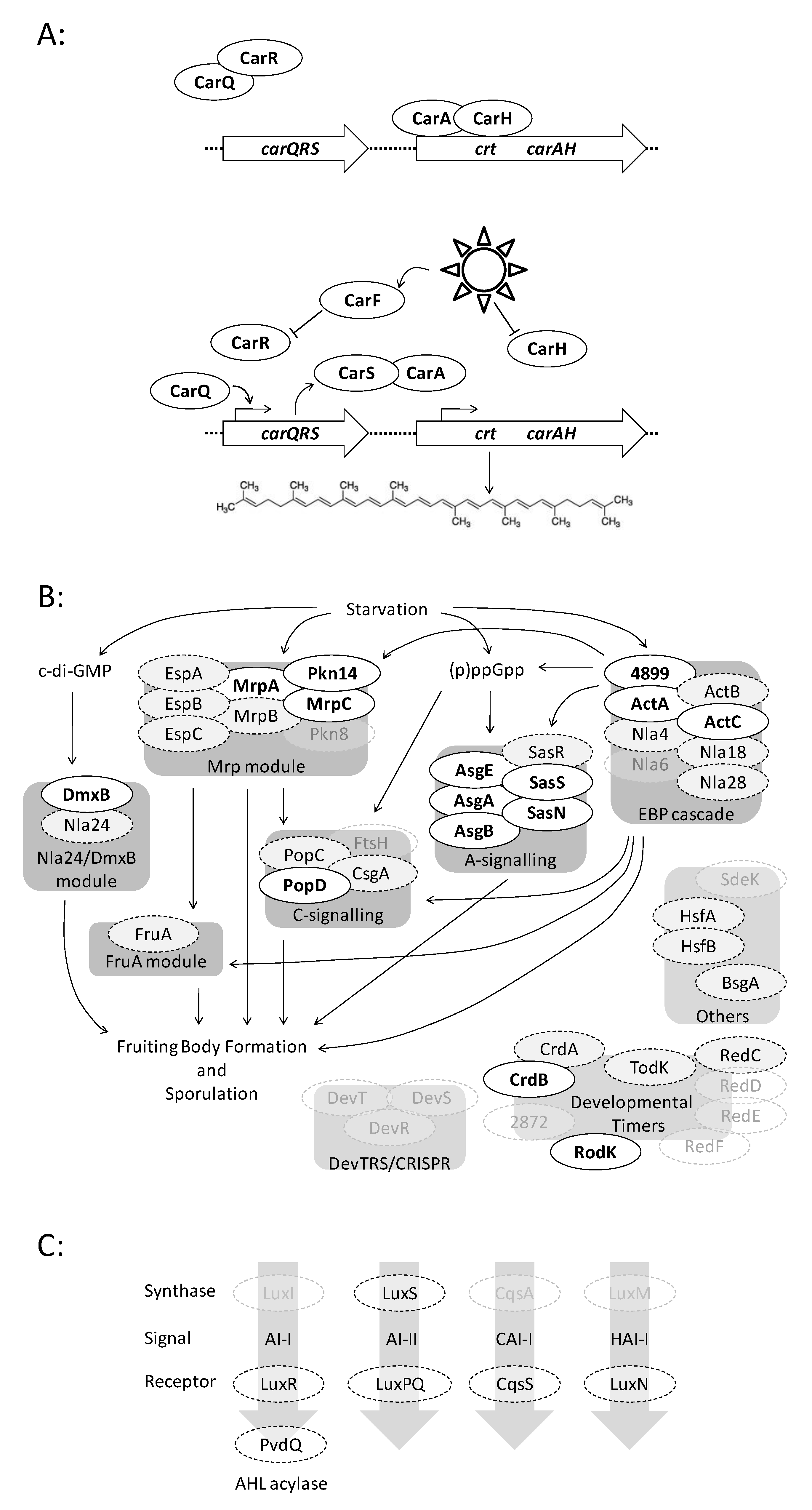
3.6. Case Study 1: Carotenogenesis
3.7. Case Study 2: Fruiting Body Formation
3.8. Case Study 3: Quorum Signalling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pérez, J.; Castañeda-García, A.; Jenke-Kodama, H.; Müller, R.; Muñoz-Dorado, J. Eukaryotic-like protein kinases in the prokaryotes and the myxobacterial kinome. Proc. Natl. Acad. Sci. USA 2008, 105, 15950–15955. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, D.E.; Cock, P.J.A. Two-component systems of the myxobacteria: Structure, diversity and evolutionary relationships. Microbiology 2008, 154 Pt 2, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, D.E. Genome-wide analysis of myxobacterial two-component systems: Genome relatedness and evolutionary changes. BMC Genom. 2015, 16, 780. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Gerth, K.; Stadler, M.; Dogma, I.J., Jr.; Müller, R. Expanded phylogeny of myxobacteria and evidence for cultivation of the ‘unculturables’. Mol. Phylogenet. Evol. 2010, 57, 878–887. [Google Scholar] [CrossRef]
- Sharma, G.; Narwani, T.; Subramanian, S. Complete Genome Sequence and Comparative Genomics of a Novel Myxobacterium Myxococcus hansupus. PLoS ONE 2016, 11, e0148593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, P.G.; Ingleby, O.; Girdwood, S.; Cookson, A.R.; Morphew, R.M.; Whitworth, D.E. Predatory Organisms with Untapped Biosynthetic Potential: Descriptions of Novel Corallococcus Species C. aberystwythensis sp. nov., C. carmarthensis sp. nov., C. exercitus sp. nov., C. interemptor sp. nov., C. llansteffanensis sp. nov., C. praedator sp. nov., C. sicarius sp. nov., and C. terminator sp. nov. Appl. Environ. Microbiol. 2020, 86, e01931-19. [Google Scholar] [PubMed]
- Chambers, J.; Sparks, N.; Sydney, N.; Livingstone, P.G.; Cookson, A.R.; Whitworth, D.E. Comparative genomics and pan-genomics of the Myxococcaceae, including a description of five novel species: Myxococcus eversor sp. nov., Myxococcus llanfairpwllgwyngyllgogerychwyrndrobwllllantysiliogogogochensis sp. nov., Myxococcus vastator sp. nov., Pyxidicoccus caerfyrddinensis sp. nov., and Pyxidicoccus trucidator sp. nov. Genome Biol. Evol. 2020, in press. [Google Scholar]
- Livingstone, P.G.; Morphew, R.M.; Whitworth, D.E. Genome Sequencing and Pan-Genome Analysis of 23 Corallococcus spp. Strains Reveal Unexpected Diversity, with Particular Plasticity of Predatory Gene Sets. Front. Microbiol. 2018, 9, 3187. [Google Scholar] [CrossRef] [Green Version]
- Zwarycz, A.S.; Livingstone, P.G.; Whitworth, D.E. Within-species variation in OMV cargo proteins: The Myxococcus xanthus OMV pan-proteome. Mol. Omics 2020. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Cameron Thrash, J.; Temperton, B. Implications of streamlining theory for microbial ecology. ISME J. 2014, 8, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Sutton, D.; Livingstone, P.G.; Furness, E.; Swain, M.T.; Whitworth, D.E. Genome-Wide Identification of Myxobacterial Predation Genes and Demonstration of Formaldehyde Secretion as a Potentially Predation-Resistant Trait of Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2650. [Google Scholar] [CrossRef]
- Galbis-Martínez, M.; Padmanabhan, S.; Murillo, F.J.; Elías-Arnanz, M. CarF mediates signaling by singlet oxygen, generated via photoexcited protoporphyrin IX in Myxococcus xanthus light-induced carotenogenesis. J. Bacteriol. 2012, 194, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Browning, D.F.; Whitworth, D.E.; Hodgson, D.A. Light-induced carotenogenesis in Myxococcus xanthus: Functional characterization of the ECF sigma factor CarQ and antisigma factor CarR. Mol. Microbiol. 2003, 48, 237–251. [Google Scholar] [CrossRef]
- Whitworth, D.E.; Bryan, S.J.; Berry, A.E.; McGowan, S.J.; Hodgson, D.A. Genetic dissection of the light-inducible carQRS promoter region of Myxococcus xanthus. J. Bacteriol. 2004, 186, 7836–7846. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, D.E.; Hodgson, D.A. Light-induced carotenogenesis in Myxococcus xanthus: Evidence that CarS acts as an anti-repressor of CarA. Mol. Microbiol. 2001, 42, 809–819. [Google Scholar] [CrossRef]
- López-Rubio, J.J.; Elías-Arnanz, M.; Padmanabhan, S.; Murillo, F.J. A repressor-antirepressor pair links two loci controlling light-induced carotenogenesis in Myxococcus xanthus. J. Biol. Chem. 2002, 277, 7262–7270. [Google Scholar] [CrossRef] [Green Version]
- León, E.; Navarro-Avilés, G.; Santiveri, C.M.; Flores-Flores, C.; Rico, M.; González, C.; Murillo, F.J.; Elías-Arnanz, M.; Jiménez, M.A.; Padmanabhan, S. A bacterial antirepressor with SH3 domain topology mimics operator DNA in sequestering the repressor DNA recognition helix. Nucleic Acids Res. 2010, 38, 5226–5241. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Jost, M.; Drennan, C.L.; Elías-Arnanz, M. A New Facet of Vitamin B(12): Gene Regulation by Cobalamin-Based Photoreceptors. Annu. Rev. Biochem. 2017, 86, 485–514. [Google Scholar] [CrossRef]
- Sarwar, Z.; Garza, A.G. Two-Component Signal Transduction Systems That Regulate the Temporal and Spatial Expression of Myxococcus xanthus Sporulation Genes. J. Bacteriol. 2015, 198, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Bretl, D.J.; Kirby, J.R. Molecular Mechanisms of Signaling in Myxococcus xanthus Development. J. Mol. Biol. 2016, 428, 3805–3830. [Google Scholar] [CrossRef]
- Muñoz-Dorado, J.; Moraleda-Muñoz, A.; Marcos-Torres, F.J.; Contreras-Moreno, F.J.; Martin-Cuadrado, A.B.; Schrader, J.M.; Higgs, P.I.; Pérez, J. Transcriptome dynamics of the Myxococcus xanthus multicellular developmental program. Elife 2019, 8, e50374. [Google Scholar] [CrossRef]
- Kroos, L. Highly Signal-Responsive Gene Regulatory Network Governing Myxococcus Development. Trends Genet. 2017, 33, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Konovalova, A.; Wegener-Feldbrügge, S.; Søgaard-Andersen, L. Two intercellular signals required for fruiting body formation in Myxococcus xanthus act sequentially but non-hierarchically. Mol. Microbiol. 2012, 86, 65–81. [Google Scholar] [CrossRef]
- Ng, W.L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Lloyd, D.G.; Whitworth, D.E. The Myxobacterium Myxococcus xanthus Can Sense and Respond to the Quorum Signals Secreted by Potential Prey Organisms. Front. Microbiol. 2017, 8, 439. [Google Scholar] [CrossRef]
- Whitworth, D.E.; Swain, M.T. A survey of non-coding RNAs in the social and predatory myxobacterium Myxococcus xanthus DK1622. Mol. Omics 2020. [Google Scholar] [CrossRef]
- Livingstone, P.G.; Morphew, R.M.; Whitworth, D.E. Myxobacteria Are Able to Prey Broadly upon Clinically-Relevant Pathogens, Exhibiting a Prey Range Which Cannot Be Explained by Phylogeny. Front. Microbiol. 2017, 8, 1593. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef]
- Barakat, M.; Ortet, P.; Whitworth, D.E. P2RP: A Web-based framework for the identification and analysis of regulatory proteins in prokaryotic genomes. BMC Genom. 2013, 14, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortet, P.; De Luca, G.; Whitworth, D.E.; Barakat, M. P2TF: A comprehensive resource for analysis of prokaryotic transcription factors. BMC Genom. 2012, 13, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortet, P.; Whitworth, D.E.; Santaella, C.; Achouak, W.; Barakat, M. P2CS: Updates of the prokaryotic two-component systems database. Nucleic Acids Res. 2015, 43, D536–D541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, B.S.; Nierman, W.C.; Kaiser, D.; Slater, S.C.; Durkin, A.S.; Eisen, J.A.; Ronning, C.M.; Barbazuk, W.B.; Blanchard, M.; Field, C.; et al. Evolution of sensory complexity recorded in a myxobacterial genome. Proc. Natl. Acad. Sci. USA 2006, 103, 15200–15205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treuner-Lange, A.; Bruckskotten, M.; Rupp, O.; Goesmann, A.; Søgaard-Andersen, L. Complete Genome Sequence of the Fruiting Myxobacterium Myxococcus macrosporus Strain DSM 14697, Generated by PacBio Sequencing. Genome Announc. 2017, 5, e01127-17. [Google Scholar] [CrossRef] [Green Version]
- Huntley, S.; Kneip, S.; Treuner-Lange, A.; Søgaard-Andersen, L. Complete genome sequence of Myxococcus stipitatus strain DSM 14675, a fruiting myxobacterium. Genome Announc. 2013, 1, e0010013. [Google Scholar] [CrossRef] [Green Version]
- Huntley, S.; Zhang, Y.; Treuner-Lange, A.; Kneip, S.; Sensen, C.W.; Søgaard-Andersen, L. Complete genome sequence of the fruiting myxobacterium Corallococcus coralloides DSM 2259. J. Bacteriol. 2012, 194, 3012–3013. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Diodati, M.E.; Gill, R.E.; Plamann, L.; Singer, M. Initiation and early developmental events. In Myxobacteria: Multicellularity and Differentiation; Whitworth, D.E., Ed.; American Society for Microbiology: Washington, DC, USA, 2008; pp. 43–76. [Google Scholar]
- Francis, V.I.; Porter, S.L. Multikinase Networks: Two-Component Signaling Networks Integrating Multiple Stimuli. Annu. Rev. Microbiol. 2019, 73, 199–223. [Google Scholar] [CrossRef]
- Payne, J.L.; Wagner, A. Mechanisms of mutational robustness in transcriptional regulation. Front. Genet. 2014, 6, 322. [Google Scholar] [CrossRef] [Green Version]
- Silva-Rocha, R.; de Lorenzo, V. Noise and robustness in prokaryotic regulatory networks. Annu. Rev. Microbiol. 2010, 64, 257–275. [Google Scholar] [CrossRef]
- Agarwal, S. Systems approaches in understanding evolution and evolvability. Prog. Biophys. Mol. Biol. 2013, 113, 369–374. [Google Scholar] [CrossRef]
- Yu, Y.T.; Yuan, X.; Velicer, G.J. Adaptive evolution of ansRNA that controls Myxococcus development. Science 2010, 328, 993. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.N.; Kleiner, M.; Velicer, G.J. Spontaneous Reversions of an Evolutionary Trait Loss Reveal Regulators of a Small RNA That Controls Multicellular Development in Myxobacteria. J. Bacteriol. 2016, 198, 3142–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusick, J.K.; Hager, E.; Gill, R.E. Identification of a mutant locus that bypasses the BsgA protease requirement for social development in Myxococcus xanthus. FEMS Microbiol. Lett. 2015, 362, 1–8. [Google Scholar] [CrossRef]
- Goldman, B.; Bhat, S.; Shimkets, L.J. Genome evolution and the emergence of fruiting body development in Myxococcus xanthus. PLoS ONE 2007, 2, e1329. [Google Scholar] [CrossRef] [Green Version]
- Albataineh, H.; Duke, M.; Misra, S.K.; Sharp, J.S.; Cole Stevens, D. Cryptic, solo acyl homoserine lactone synthase from predatory myxobacterium suggests beneficial contribution to prey quorum signaling. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Henke, J.M.; Bassler, B.L. Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J. Bacteriol. 2004, 186, 6902–6914. [Google Scholar] [CrossRef] [Green Version]
- Polkade, A.V.; Mantri, S.S.; Patwekar, U.J.; Jangid, K. Quorum Sensing: An Under-Explored Phenomenon in the Phylum Actinobacteria. Front. Microbiol. 2016, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, D.E. Genomes and knowledge—A questionable relationship? Trends Microbiol. 2008, 16, 512–519. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A: | ||||||||
| Genus | Species | Strain | Contigs | Size (Mbp) | % GC | CDS | Accession | Reference |
| Myxococcus | xanthus | AB022 | 257 | 9.06 | 68.9 | 6995 | VHLD00000000 | [11] |
| Myxococcus | xanthus | AB024B | 365 | 9.06 | 68.9 | 7013 | SRLY00000000 | [11] |
| Myxococcus | xanthus | AB056 | 230 | 9.11 | 69.1 | 7065 | VHLB00000000 | [11] |
| Myxococcus | xanthus | CA005 | 227 | 9.11 | 68.9 | 7175 | SRLV00000000 | [11] |
| Myxococcus | xanthus | CA006 | 360 | 9.05 | 68.9 | 6991 | SRLU00000000 | [11] |
| Myxococcus | xanthus | CA010 | 250 | 9.05 | 68.9 | 6981 | VHLA00000000 | [11] |
| Myxococcus | xanthus | CA018 | 727 | 9.07 | 68.8 | 7102 | JAAEAG000000000 | [9] |
| Myxococcus | xanthus | CA023 | 235 | 9.08 | 68.9 | 7047 | JAAEAH000000000 | [9] |
| Myxococcus | xanthus | CA027 | 252 | 9.05 | 68.9 | 7001 | WBSK00000000 | [9] |
| Myxococcus | xanthus | DK1622 | 1 | 9.14 | 68.9 | 7216 | GCF_000012685 | [36] |
| 9.08 ± 0.03 | 68.9 ± 0.1 | 7059 ± 82 | ||||||
| B: | ||||||||
| Genus | Species | Strain | Contigs | Size (Mbp) | % GC | CDS | Accession | Reference |
| Myxococcus | xanthus/virescens | DSM 2260 | 57 | 9.24 | 69.2 | 7340 | FNAJ00000000 | JGI |
| Myxococcus | eversor | AB053B | 124 | 11.39 | 68.9 | 8751 | JAAIXY01000000000 | [7] |
| Myxococcus | fulvus | DSM 16525 | 42 | 10.82 | 70.0 | 8318 | FOIB00000000 | JGI |
| Myxococcus | hansupus | Mixupus | 1 | 9.49 | 69.2 | 7069 | GCA_000280925 | [5] |
| Myxococcus | llanfairPGensis | AM401 | 1077 | 12.41 | 68.7 | 9508 | VIFM00000000 | [7] |
| Myxococcus | macrosporus | DSM 14697 | 1 | 8.97 | 70.6 | 6966 | GCA_002305895 | [37] |
| Myxococcus | stipitatus | DSM 14675 | 1 | 10.35 | 69.2 | 7796 | GCA_000331735 | [38] |
| Myxococcus | vastator | AM301 | 1008 | 8.99 | 69.9 | 7055 | JAAIYB000000000 | [7] |
| Pyxidicoccus | caerfyrddinensis | CA032A | 177 | 12.67 | 70.2 | 9986 | JAAIYA000000000 | [7] |
| Pyxidicoccus | fallax | DSM 14698 | 825 | 13.53 | 70.5 | 10513 | JABBJJ000000000 | [7] |
| Pyxidicoccus | trucidator | CA060A | 136 | 12.67 | 70.3 | 9355 | JAAIXZ000000000 | [7] |
| 10.96 ± 1.68 | 69.7 ± 0.6 | 8423 ± 1279 | ||||||
| C: | ||||||||
| Genus | Species | Strain | Contigs | Size (Mbp) | % GC | CDS | Accession | Reference |
| Corallococcus | aberyswythensis | AB050A | 625 | 9.98 | 70.0 | 7905 | RAWK00000000 | [8] |
| Corallococcus | carmarthensis | CA043D | 530 | 10.79 | 69.9 | 8511 | RAWE00000000 | [8] |
| Corallococcus | coralloides | DSM 2259 | 1 | 10.08 | 69.9 | 7893 | GCA_000255295 | [39] |
| Corallococcus | exercitus | AB043A | 961 | 10.15 | 70.3 | 8018 | RAVW00000000 | [8] |
| Corallococcus | interemptor | AB047A | 459 | 9.47 | 70.0 | 7566 | RAWM00000000 | [8] |
| Corallococcus | llansteffanensis | CA051B | 1244 | 10.53 | 70.3 | 8137 | RAWB00000000 | [8] |
| Corallococcus | praedator | CA031B | 1491 | 10.51 | 69.7 | 8167 | RAWI00000000 | [8] |
| Corallococcus | sicarius | CA040B | 802 | 10.39 | 70.2 | 7877 | RAWG00000000 | [8] |
| Corallococcus | terminator | CA054A | 863 | 10.35 | 69.5 | 8008 | RAVZ00000000 | [8] |
| Corallococcus | exiguus | DSM 14696 | 36 | 10.41 | 69.6 | 8112 | JAAAPK000000000 | [8] |
| 10.27 ± 0.37 | 69.9 ± 0.3 | 8019 ± 245 | ||||||
| D: | ||||||||
| Genus | Species | Strain | Contigs | Size (Mbp) | % GC | CDS | Accession | Reference |
| Corallococcus | exiguus | AB004 | 735 | 10.60 | 69.4 | 8223 | RAWS00000000 | [8] |
| Corallococcus | exiguus | AB016 | 1212 | 10.75 | 69.6 | 8940 | JABEKY000000000 | This Study |
| Corallococcus | exiguus | AB018 | 647 | 10.45 | 69.4 | 8185 | RAWR00000000 | [8] |
| Corallococcus | exiguus | AB030 | 552 | 10.63 | 69.6 | 8334 | RAWQ00000000 | [8] |
| Corallococcus | exiguus | AB031 | 611 | 10.43 | 69.7 | 8356 | JABEKZ000000000 | This Study |
| Corallococcus | exiguus | AB032C | 298 | 10.45 | 69.5 | 8078 | RAWP00000000 | [8] |
| Corallococcus | exiguus | AB038B | 471 | 10.77 | 69.3 | 8409 | RAWO00000000 | [8] |
| Corallococcus | exiguus | AB051 | 1378 | 10.76 | 69.6 | 9025 | JABELA000000000 | This Study |
| Corallococcus | exiguus | CA041A | 794 | 10.26 | 69.5 | 8071 | RAWF00000000 | [8] |
| Corallococcus | exiguus | CA048 | 723 | 10.35 | 69.6 | 8428 | JABELB000000000 | This Study |
| 10.55 ± 0.18 | 69.5 ± 0.1 | 8405 ± 330 |
| M. xanthus Strains (10) | Myxococcus/Pyxidicoccus spp. (11) | Corallococcus spp. (10) | C. exiguus Strains (10) | |||||
|---|---|---|---|---|---|---|---|---|
| (mean ± sd) | (% Variability) | (mean ± sd) | (% Variability) | (mean ± sd) | (% Variability) | (mean ± sd) | (% Variability) | |
| TCS | 280 ± 2 | 0.81 | 329 ± 51 | 15.58 | 306 ± 12 | 4.05 | 302 ± 3 | 1.15 |
| HK | 141 ± 2 | 1.62 | 174 ± 35 | 19.94 | 160 ± 6 | 3.98 | 154 ± 2 | 1.00 |
| P | 3 ± 1 | 22.22 | 3 ± 1 | 33.92 | 3 ± 1 | 31.43 | 4 ± 2 | 40.9 |
| RR | 136 ± 1 | 0.90 | 151 ± 17 | 11.13 | 143 ± 7 | 4.56 | 143 ± 2 | 1.05 |
| S/T Kinases | 92 ± 5 | 5.25 | 124 ± 31 | 25.00 | 107 ± 8 | 7.00 | 113 ± 5 | 4.43 |
| TF | 263 ± 5 | 1.73 | 363 ± 79 | 21.86 | 341 ± 24 | 7.04 | 358 ± 10 | 2.89 |
| TR | 123 ± 2 | 1.33 | 171 ± 38 | 22.04 | 165 ± 11 | 6.92 | 178 ± 10 | 5.52 |
| OCS | 36 ± 3 | 7.18 | 70 ± 27 | 38.46 | 73 ± 9 | 11.58 | 71 ± 7 | 9.71 |
| RR TF | 50 ± 0 | 0.85 | 58 ± 8 | 13.42 | 48 ± 4 | 8.43 | 53 ± 1 | 1.96 |
| σ factors | 55 ± 1 | 1.77 | 65 ± 12 | 19.08 | 55 ± 3 | 5.75 | 56 ± 2 | 4.04 |
| CDS | 7059 ± 82 | 1.16 | 8423 ± 1279 | 15.18 | 8019 ± 245 | 3.06 | 8405 ± 330 | 3.93 |
| Protein Class | P | P | RR | RR | RR | RR | RR | RR | RR | RR | TR | TR | TR | TR | TR | TR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein Family | HisKA | Hpt | CheV | CyC-C | LytTR | OmpR | PrrA | TrxB | VieB | Xre | MerR | Fur | HrcA | NrdR | PucR | Rok |
| M. xanthus AB022 | 3 | 1 | 2 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus AB024B | 3 | 1 | 1 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus AB056 | 2 | 1 | 2 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus CA005 | 4 | 1 | 2 | 2 | 11 | 2 | 1 | 1 | 4 | 2 | 1 | 1 | 1 | |||
| M. xanthus CA006 | 3 | 1 | 1 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus CA010 | 3 | 1 | 1 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus CA018 | 2 | 1 | 1 | 2 | 11 | 2 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | |||
| M. xanthus CA023 | 3 | 1 | 2 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus CA027 | 3 | 1 | 2 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. xanthus DK1622 | 4 | 1 | 2 | 2 | 11 | 2 | 1 | 1 | 4 | 2 | 1 | 1 | 1 | |||
| Core (per M. xanthus strain) | 2 | 0 | 1 | 1 | 2 | 11 | 2 | 0 | 1 | 1 | 4 | 2 | 1 | 1 | 0 | 1 |
| Accessory (per M. xanthus strain) | 1 | 0 | 0 | 0.6 | 0 | 0 | 0 | 0 | 0 | 0 | 0.9 | 0 | 0 | 0 | 0 | 0 |
| M. xanthus (virescens) DSM 2260 | 2 | 1 | 1 | 2 | 11 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. eversor AB053B | 3 | 1 | 2 | 12 | 18 | 2 | 1 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |
| M. fulvus DSM 16525 | 3 | 1 | 2 | 11 | 13 | 2 | 1 | 1 | 1 | 7 | 3 | 1 | 1 | 1 | ||
| M. hansupus Mixupus | 2 | 1 | 2 | 6 | 14 | 2 | 1 | 4 | 2 | 1 | 1 | 1 | ||||
| M. llanfairPGensis AM401 | 5 | 1 | 2 | 14 | 15 | 2 | 1 | 1 | 1 | 7 | 2 | 1 | 1 | 1 | ||
| M. macrosporus DSM 14697 | 3 | 1 | 2 | 2 | 10 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| M. stipitatus DSM 14675 | 3 | 1 | 2 | 12 | 13 | 2 | 1 | 1 | 1 | 5 | 2 | 1 | 1 | |||
| M. vastator AM301 | 5 | 1 | 1 | 2 | 9 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | |||
| P. caerfyrddinensis CA032A | 3 | 1 | 2 | 10 | 14 | 2 | 1 | 1 | 7 | 2 | 1 | 1 | 1 | |||
| P. fallax DSM 14698 | 2 | 1 | 1 | 3 | 8 | 14 | 2 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | 1 | |
| P. trucidator CA060A | 3 | 1 | 2 | 10 | 13 | 2 | 1 | 7 | 2 | 1 | 1 | 1 | 1 | |||
| C. aberyswythensis AB050A | 3 | 1 | 2 | 5 | 9 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. carmarthensis CA043D | 3 | 1 | 2 | 8 | 12 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. coralloides DSM 2259 | 2 | 1 | 2 | 5 | 9 | 1 | 1 | 7 | 2 | 1 | 1 | 1 | 1 | |||
| C. exercitus AB043A | 3 | 1 | 2 | 5 | 11 | 1 | 1 | 8 | 2 | 1 | 1 | 1 | ||||
| C. interemptor AB047A | 2 | 1 | 2 | 2 | 8 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | ||||
| C. llansteffanensis CA051B | 4 | 1 | 2 | 7 | 9 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | ||||
| C. praedator CA031B | 5 | 1 | 2 | 8 | 11 | 1 | 1 | 7 | 2 | 1 | 1 | 1 | ||||
| C. sicarius CA040B | 3 | 1 | 2 | 4 | 10 | 1 | 1 | 8 | 2 | 1 | 1 | 1 | ||||
| C. terminator CA054A | 3 | 1 | 2 | 7 | 10 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | ||||
| C. exiguus DSM 14696 | 2 | 1 | 2 | 6 | 10 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | ||||
| C. exiguus AB004 | 3 | 1 | 2 | 7 | 13 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. exiguus AB016 | 8 | 1 | 2 | 7 | 13 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. exiguus AB018 | 3 | 1 | 1 | 2 | 6 | 11 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | 1 | ||
| C. exiguus AB030 | 3 | 1 | 1 | 2 | 6 | 11 | 1 | 1 | 5 | 2 | 1 | 1 | 1 | 1 | ||
| C. exiguus AB031 | 3 | 1 | 2 | 7 | 13 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. exiguus AB032C | 3 | 1 | 2 | 7 | 13 | 1 | 1 | 7 | 2 | 1 | 1 | 1 | 1 | |||
| C. exiguus AB038B | 3 | 1 | 2 | 8 | 12 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. exiguus AB051 | 5 | 1 | 2 | 7 | 13 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | |||
| C. exiguus CA041A | 2 | 1 | 1 | 2 | 5 | 11 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 | ||
| C. exiguus CA048 | 3 | 1 | 2 | 6 | 11 | 1 | 1 | 6 | 2 | 1 | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitworth, D.E.; Zwarycz, A. A Genomic Survey of Signalling in the Myxococcaceae. Microorganisms 2020, 8, 1739. https://doi.org/10.3390/microorganisms8111739
Whitworth DE, Zwarycz A. A Genomic Survey of Signalling in the Myxococcaceae. Microorganisms. 2020; 8(11):1739. https://doi.org/10.3390/microorganisms8111739
Chicago/Turabian StyleWhitworth, David E., and Allison Zwarycz. 2020. "A Genomic Survey of Signalling in the Myxococcaceae" Microorganisms 8, no. 11: 1739. https://doi.org/10.3390/microorganisms8111739
APA StyleWhitworth, D. E., & Zwarycz, A. (2020). A Genomic Survey of Signalling in the Myxococcaceae. Microorganisms, 8(11), 1739. https://doi.org/10.3390/microorganisms8111739