Bile Acid Signal Molecules Associate Temporally with Respiratory Inflammation and Microbiome Signatures in Clinically Stable Cystic Fibrosis Patients


 , ,
on behalf of AREST CF
, ,
on behalf of AREST CF
Abstract
:1. Introduction
2. Materials and Methods
2.1. BALF Cohort
2.2. Bile Acid Profiling and Bacterial DNA Extraction
2.3. Profiling and Analysis of the BALF-Associated Microbial Communities
2.4. Methodological Strategy to Minimize the Effect of Environmental Contaminants in the BALF-Associated Microbial Profiles
2.5. Statistical Analysis
2.6. Ethics, Consent and Permissions
2.7. Data Availability
3. Results
3.1. Study Cohort
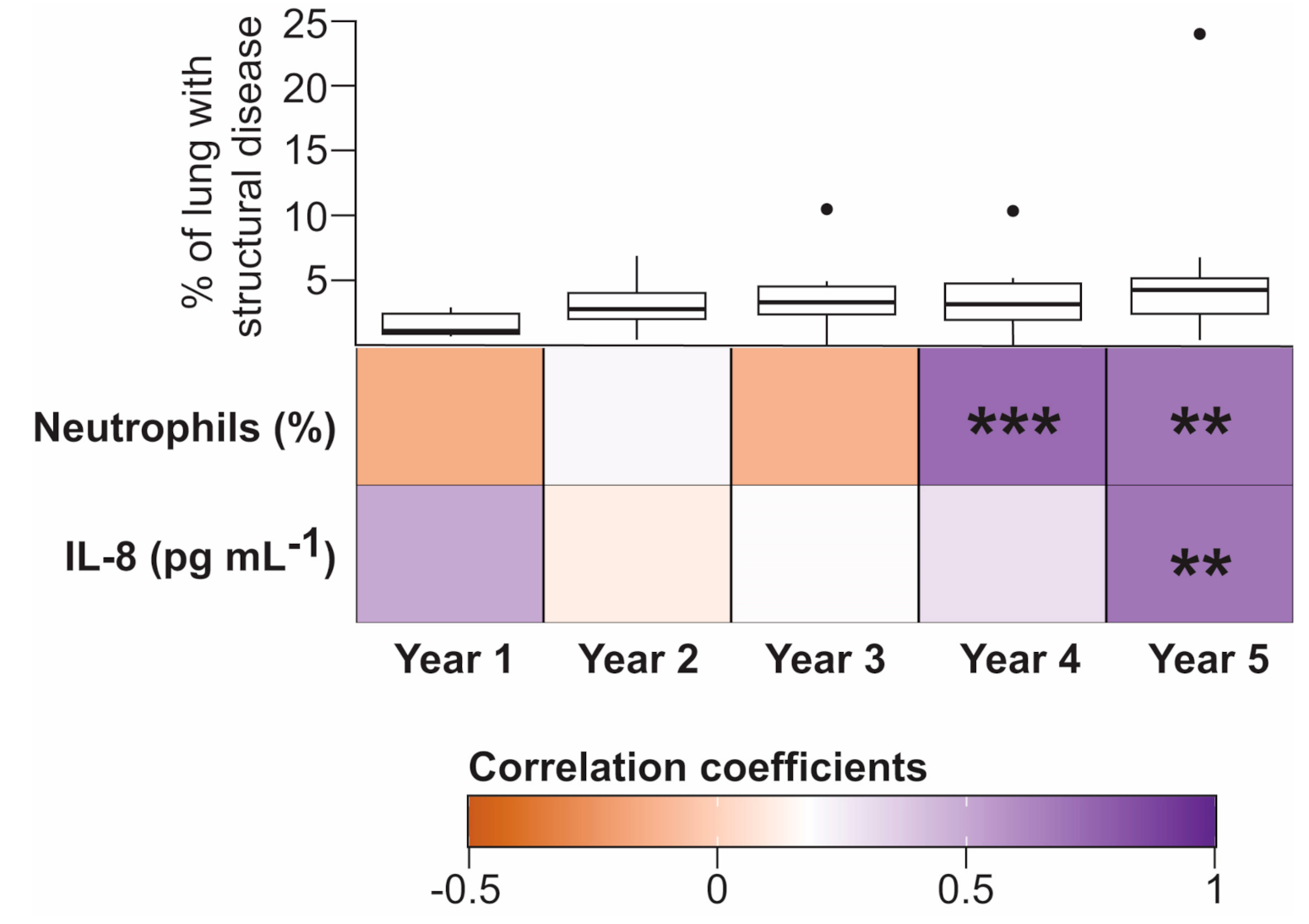
3.2. Temporal Associations between Bile Acids and Inflammatory Markers
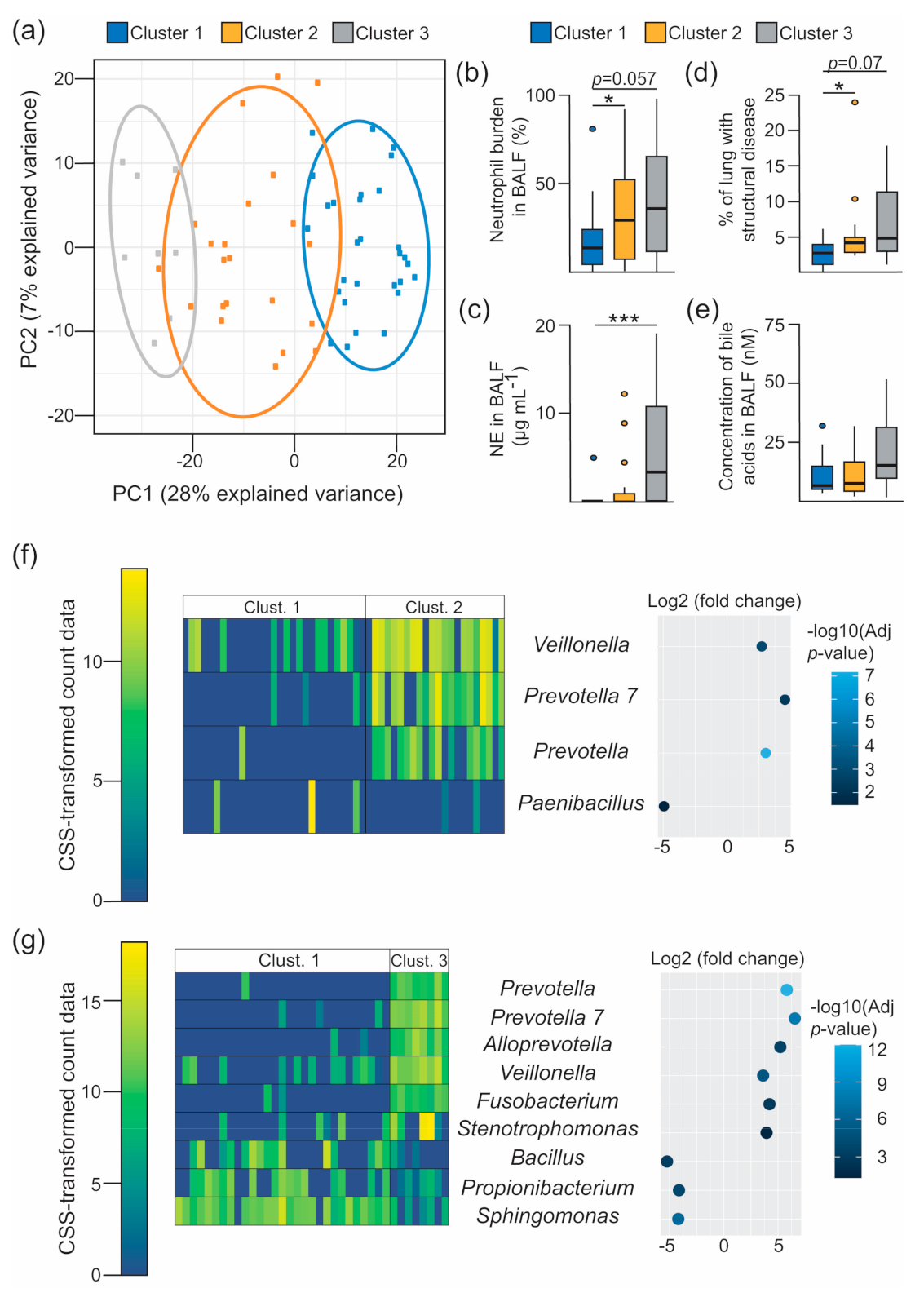
3.3. Characterisation of the 16S-Based BALF Microbial Structures and Their Association with Disease Outcomes
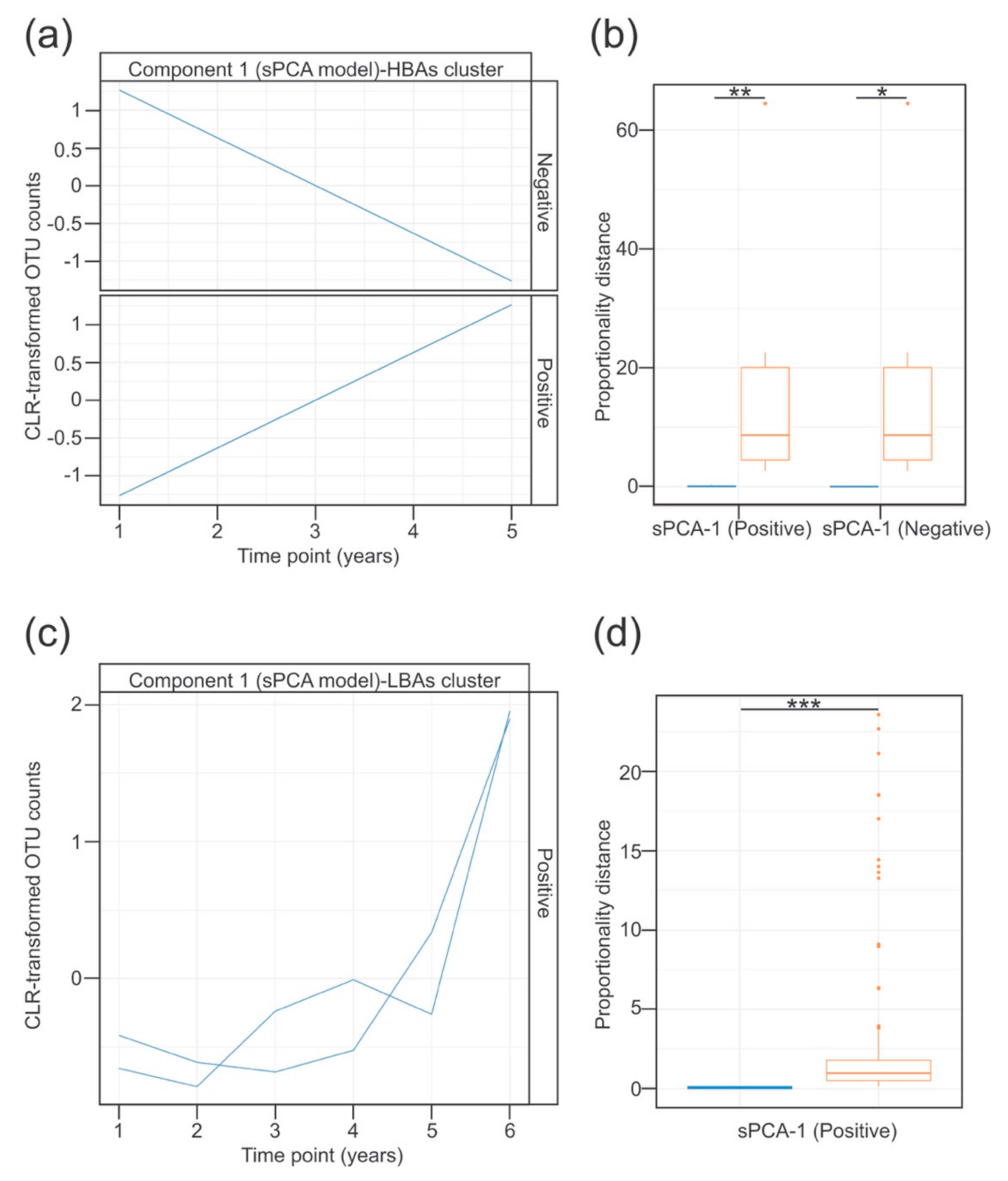
3.4. Temporal Dynamics of the Lung Microbiota Associated with the Detection of Bile Acids in BALF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Kreda, S.M.; Davis, C.W.; Rose, M.C. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb. Perspect. Med. 2012, 2, a009589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, S.C.; Hall, G.L.; Sly, P.D.; Stick, S.M.; Douglas, T.A.; Australian Respiratory Early Surveillance Team for Cystic, F. Early Lung Disease in Infants and Preschool Children with Cystic Fibrosis. What Have We Learned and What Should We Do about It? Am. J. Respir. Crit. Care Med. 2017, 195, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhlebach, M.S.; Zorn, B.T.; Esther, C.R.; Hatch, J.E.; Murray, C.P.; Turkovic, L.; Ranganathan, S.C.; Boucher, R.C.; Stick, S.M.; Wolfgang, M.C. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog. 2018, 14, e1006798. [Google Scholar] [CrossRef]
- Cuthbertson, L.; Walker, A.W.; Oliver, A.E.; Rogers, G.B.; Rivett, D.W.; Hampton, T.H.; Ashare, A.; Elborn, J.S.; De Soyza, A.; Carroll, M.P.; et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Marsland, B.J.; Trompette, A.; Gollwitzer, E.S. The Gut-Lung Axis in Respiratory Disease. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 2), S150–S156. [Google Scholar] [CrossRef]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut-lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef]
- Anderson, J.L.; Miles, C.; Tierney, A.C. Effect of probiotics on respiratory, gastrointestinal and nutritional outcomes in patients with cystic fibrosis: A systematic review. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2017, 16, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Falkowski, N.R.; Huffnagle, G.B.; Curtis, J.L. Bacterial Topography of the Healthy Human Lower Respiratory Tract. mBio 2017, 8. [Google Scholar] [CrossRef]
- Reen, F.J.; Woods, D.F.; Mooij, M.J.; Adams, C.; O’Gara, F. Respiratory pathogens adopt a chronic lifestyle in response to bile. PLoS ONE 2012, 7, e45978. [Google Scholar] [CrossRef]
- Reen, F.J.; Flynn, S.; Woods, D.F.; Dunphy, N.; Chroinin, M.N.; Mullane, D.; Stick, S.; Adams, C.; O’Gara, F. Bile signalling promotes chronic respiratory infections and antibiotic tolerance. Sci. Rep. 2016, 6, 29768. [Google Scholar] [CrossRef] [Green Version]
- Caparros-Martin, J.A.; Flynn, S.; Reen, F.J.; Woods, D.F.; Agudelo-Romero, P.; Ranganathan, S.C.; Stick, S.M.; O’Gara, F. The Detection of Bile Acids in the Lungs of Paediatric Cystic Fibrosis Patients Is Associated with Altered Inflammatory Patterns. Diagnostics 2020, 10, 282. [Google Scholar] [CrossRef]
- Caparros-Martin, J.A.; Lareu, R.R.; Ramsay, J.P.; Peplies, J.; Reen, F.J.; Headlam, H.A.; Ward, N.C.; Croft, K.D.; Newsholme, P.; Hughes, J.D.; et al. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome 2017, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Le Cao, K.A.; Costello, M.E.; Lakis, V.A.; Bartolo, F.; Chua, X.Y.; Brazeilles, R.; Rondeau, P. MixMC: A Multivariate Statistical Framework to Gain Insight into Microbial Communities. PLoS ONE 2016, 11, e0160169. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- De Goffau, M.C.; Lager, S.; Sovio, U.; Gaccioli, F.; Cook, E.; Peacock, S.J.; Parkhill, J.; Charnock-Jones, D.S.; Smith, G.C.S. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–334. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Goeman, J.J.; van de Geer, S.A.; de Kort, F.; van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Bodein, A.; Chapleur, O.; Droit, A.; Le Cao, K.A. A Generic Multivariate Framework for the Integration of Microbiome Longitudinal Studies with Other Data Types. Front. Genet. 2019, 10, 963. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Barter, R.; Yu, B. Superheat: A Graphical Tool for Exploring Complex Datasets Using Heatmaps. R Package Version 0.1.0. 2017. Available online: https://CRAN.R-project.org/package=superheat (accessed on 1 June 2020).
- Kassambara, A.; Mundt, F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses. 2020. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 1 June 2020).
- Lüdecke, D. sjPlot: Data Visualization for Statistics in Social Science. 2020. Available online: https://CRAN.R-project.org/package=sjPlot (accessed on 1 September 2020).
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Rosenow, T.; Oudraad, M.C.; Murray, C.P.; Turkovic, L.; Kuo, W.; de Bruijne, M.; Ranganathan, S.C.; Tiddens, H.A.; Stick, S.M.; Australian Respiratory Early Surveillance Team for Cystic, F. PRAGMA-CF. A Quantitative Structural Lung Disease Computed Tomography Outcome in Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 191, 1158–1165. [Google Scholar] [CrossRef]
- Sly, P.D.; Brennan, S.; Gangell, C.; de Klerk, N.; Murray, C.; Mott, L.; Stick, S.M.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 2009, 180, 146–152. [Google Scholar] [CrossRef]
- Mott, L.S.; Park, J.; Murray, C.P.; Gangell, C.L.; de Klerk, N.H.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; Sly, P.D.; Stick, S.M.; et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 2012, 67, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. [Google Scholar] [CrossRef]
- Aldhahrani, A.; Verdon, B.; Ward, C.; Pearson, J. Effects of bile acids on human airway epithelial cells: Implications for aerodigestive diseases. ERJ Open Res. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Blondeau, K.; Dupont, L.J.; Mertens, V.; Verleden, G.; Malfroot, A.; Vandenplas, Y.; Hauser, B.; Sifrim, D. Gastro-oesophageal reflux and aspiration of gastric contents in adult patients with cystic fibrosis. Gut 2008, 57, 1049–1055. [Google Scholar] [CrossRef]
- Legendre, C.; Reen, F.J.; Woods, D.F.; Mooij, M.J.; Adams, C.; O’Gara, F. Bile acids repress hypoxia-inducible factor 1 signaling and modulate the airway immune response. Infect. Immun. 2014, 82, 3531–3541. [Google Scholar] [CrossRef] [Green Version]
- Pauwels, A.; Decraene, A.; Blondeau, K.; Mertens, V.; Farre, R.; Proesmans, M.; Van Bleyenbergh, P.; Sifrim, D.; Dupont, L.J. Bile acids in sputum and increased airway inflammation in patients with cystic fibrosis. Chest 2012, 141, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Downey, D.G.; Bell, S.C.; Elborn, J.S. Neutrophils in cystic fibrosis. Thorax 2009, 64, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Dakin, C.J.; Numa, A.H.; Wang, H.; Morton, J.R.; Vertzyas, C.C.; Henry, R.L. Inflammation, infection, and pulmonary function in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2002, 165, 904–910. [Google Scholar] [CrossRef]
- Frayman, K.B.; Armstrong, D.S.; Carzino, R.; Ferkol, T.W.; Grimwood, K.; Storch, G.A.; Teo, S.M.; Wylie, K.M.; Ranganathan, S.C. The lower airway microbiota in early cystic fibrosis lung disease: A longitudinal analysis. Thorax 2017, 72, 1104–1112. [Google Scholar] [CrossRef]
- Prevaes, S.M.; de Steenhuijsen Piters, W.A.; de Winter-de Groot, K.M.; Janssens, H.M.; Tramper-Stranders, G.A.; Chu, M.L.; Tiddens, H.A.; van Westreenen, M.; van der Ent, C.K.; Sanders, E.A.; et al. Concordance between upper and lower airway microbiota in infants with cystic fibrosis. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [Green Version]
- Marsh, R.L.; Kaestli, M.; Chang, A.B.; Binks, M.J.; Pope, C.E.; Hoffman, L.R.; Smith-Vaughan, H.C. The microbiota in bronchoalveolar lavage from young children with chronic lung disease includes taxa present in both the oropharynx and nasopharynx. Microbiome 2016, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Segal, L.N.; Alekseyenko, A.V.; Clemente, J.C.; Kulkarni, R.; Wu, B.; Gao, Z.; Chen, H.; Berger, K.I.; Goldring, R.M.; Rom, W.N.; et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome 2013, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Segal, L.N.; Clemente, J.C.; Tsay, J.C.; Koralov, S.B.; Keller, B.C.; Wu, B.G.; Li, Y.; Shen, N.; Ghedin, E.; Morris, A.; et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 2016, 1, 16031. [Google Scholar] [CrossRef] [Green Version]
- Parkins, M.D.; Floto, R.A. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2015, 14, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung Infections Associated with Cystic Fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef] [Green Version]
- Sutter, V.L. Anaerobes as normal flora. Rev. Infect. Dis. 1984, 6, S62–S66. [Google Scholar] [CrossRef]
- Espinoza, J.; Harkins, D.; Torralba, M.; Gomez, A.; Highlander, S.; Jones, M.; Leong, P.; Saffery, R.; Bockmann, M.; Kuelbs, C.; et al. Supragingival Plaque Microbiome Ecology and Functional Potential in the Context of Health and Disease. mBio 2018, 9, e01631-18. [Google Scholar] [CrossRef] [Green Version]
- Aas, J.A.; Paster, B.J.; Stokes, L.N.; Olsen, I.; Dewhirst, F.E. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 2005, 43, 5721–5732. [Google Scholar] [CrossRef] [Green Version]
- Makki, K.; Deehan, E.C.; Walter, J.; Backhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Zheng, X.; Huang, F.; Zhao, A.; Lei, S.; Zhang, Y.; Xie, G.; Chen, T.; Qu, C.; Rajani, C.; Dong, B.; et al. Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice. BMC Biol. 2017, 15, 120. [Google Scholar] [CrossRef] [Green Version]
- Aseeri, A.; Brodlie, M.; Lordan, J.; Corris, P.; Pearson, J.; Ward, C.; Manning, N. Bile acids are present in the lower airways of people with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 185, 463. [Google Scholar] [CrossRef]
- Reen, F.J.; Woods, D.F.; Mooij, M.J.; Chroinin, M.N.; Mullane, D.; Zhou, L.; Quille, J.; Fitzpatrick, D.; Glennon, J.D.; McGlacken, G.P.; et al. Aspirated bile: A major host trigger modulating respiratory pathogen colonisation in cystic fibrosis patients. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- McNally, P.; Ervine, E.; Shields, M.D.; Dimitrov, B.D.; El Nazir, B.; Taggart, C.C.; Greene, C.M.; McElvaney, N.G.; Greally, P. High concentrations of pepsin in bronchoalveolar lavage fluid from children with cystic fibrosis are associated with high interleukin-8 concentrations. Thorax 2011, 66, 140–143. [Google Scholar] [CrossRef] [Green Version]
- Clarke, D.; Gorman, I.; Ringholz, F.; McDermott, M.; Cox, D.W.; Greally, P.; Linnane, B.; Mc Nally, P. Pulmonary aspiration in preschool children with cystic fibrosis. Respir. Res. 2018, 19, 255. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef]
- Jorth, P.; Ehsan, Z.; Rezayat, A.; Caldwell, E.; Pope, C.; Brewington, J.J.; Goss, C.H.; Benscoter, D.; Clancy, J.P.; Singh, P.K. Direct Lung Sampling Indicates That Established Pathogens Dominate Early Infections in Children with Cystic Fibrosis. Cell Rep. 2019, 27, 1190–1204.e1193. [Google Scholar] [CrossRef] [Green Version]
- Zemanick, E.T.; Wagner, B.D.; Robertson, C.E.; Ahrens, R.C.; Chmiel, J.F.; Clancy, J.P.; Gibson, R.L.; Harris, W.T.; Kurland, G.; Laguna, T.A.; et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Flynn, J.M.; Niccum, D.; Dunitz, J.M.; Hunter, R.C. Evidence and Role for Bacterial Mucin Degradation in Cystic Fibrosis Airway Disease. PLoS Pathog. 2016, 12, e1005846. [Google Scholar] [CrossRef] [Green Version]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Ulluwishewa, D.; Wang, L.; Pereira, C.; Flynn, S.; Cain, E.; Stick, S.; Reen, F.J.; Ramsay, J.P.; O’Gara, F. Dissecting the regulation of bile-induced biofilm formation in Staphylococcus aureus. Microbiology 2016, 162, 1398–1406. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Spearman’s ρ | p-value | Adjusted p-Value |
|---|---|---|---|
| Log10(Bile acid concentration) (µM) | 0.339976 | 0.008425 | 0.02074 |
| Interleukin 8 (pg mL−1) | 0.216353 | 0.099796 | 0.12474 |
| Neutrophil Elastase (ng mL−1) | 0.508734 | 3.886 × 10−5 | 0.00019 |
| Neutrophils burden (%) | 0.170807 | 0.199858 | 0.19985 |
| Dis (%) | 0.38695 | 0.012444 | 0.02074 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flynn, S.; Reen, F.J.; Caparrós-Martín, J.A.; Woods, D.F.; Peplies, J.; Ranganathan, S.C.; Stick, S.M.; O'Gara, F., on behalf of AREST CF. Bile Acid Signal Molecules Associate Temporally with Respiratory Inflammation and Microbiome Signatures in Clinically Stable Cystic Fibrosis Patients. Microorganisms 2020, 8, 1741. https://doi.org/10.3390/microorganisms8111741
Flynn S, Reen FJ, Caparrós-Martín JA, Woods DF, Peplies J, Ranganathan SC, Stick SM, O'Gara F on behalf of AREST CF. Bile Acid Signal Molecules Associate Temporally with Respiratory Inflammation and Microbiome Signatures in Clinically Stable Cystic Fibrosis Patients. Microorganisms. 2020; 8(11):1741. https://doi.org/10.3390/microorganisms8111741
Chicago/Turabian StyleFlynn, Stephanie, F. Jerry Reen, Jose A. Caparrós-Martín, David F. Woods, Jörg Peplies, Sarath C. Ranganathan, Stephen M. Stick, and Fergal O'Gara on behalf of AREST CF. 2020. "Bile Acid Signal Molecules Associate Temporally with Respiratory Inflammation and Microbiome Signatures in Clinically Stable Cystic Fibrosis Patients" Microorganisms 8, no. 11: 1741. https://doi.org/10.3390/microorganisms8111741
APA StyleFlynn, S., Reen, F. J., Caparrós-Martín, J. A., Woods, D. F., Peplies, J., Ranganathan, S. C., Stick, S. M., & O'Gara, F., on behalf of AREST CF. (2020). Bile Acid Signal Molecules Associate Temporally with Respiratory Inflammation and Microbiome Signatures in Clinically Stable Cystic Fibrosis Patients. Microorganisms, 8(11), 1741. https://doi.org/10.3390/microorganisms8111741