T Cell Immunity and the Quest for Protective Vaccines against Staphylococcus aureus Infection


Abstract
:1. Introduction
2. Previous and Ongoing Human S. aureus Vaccine Trials
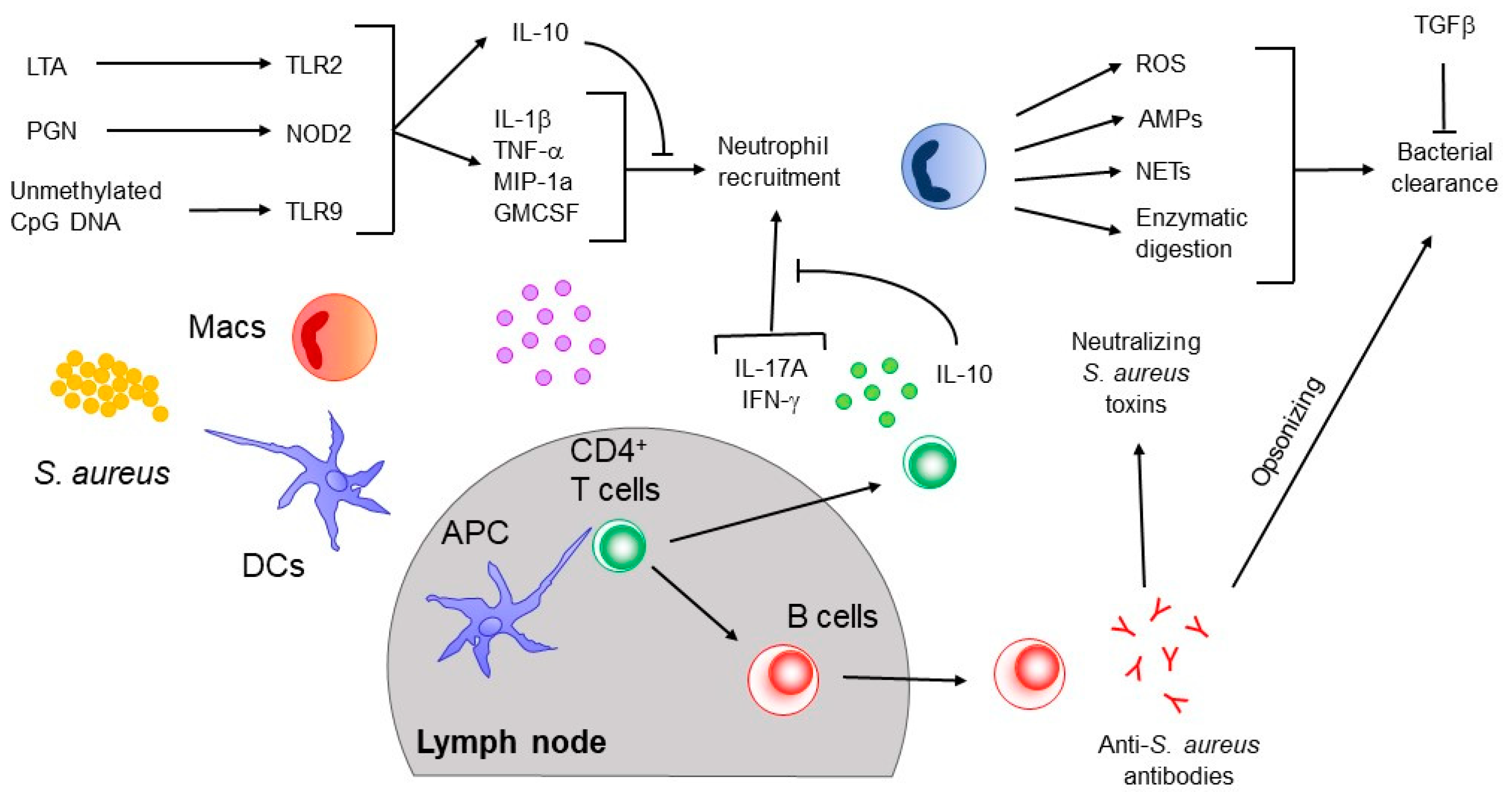
3. Innate Immune Responses to S. aureus
4. Adaptive Immune Response to S. aureus
4.1. B Cell Responses
4.2. T Cell Responses
4.2.1. Roles of γδ T Cells
4.2.2. Roles of αβ T Cells
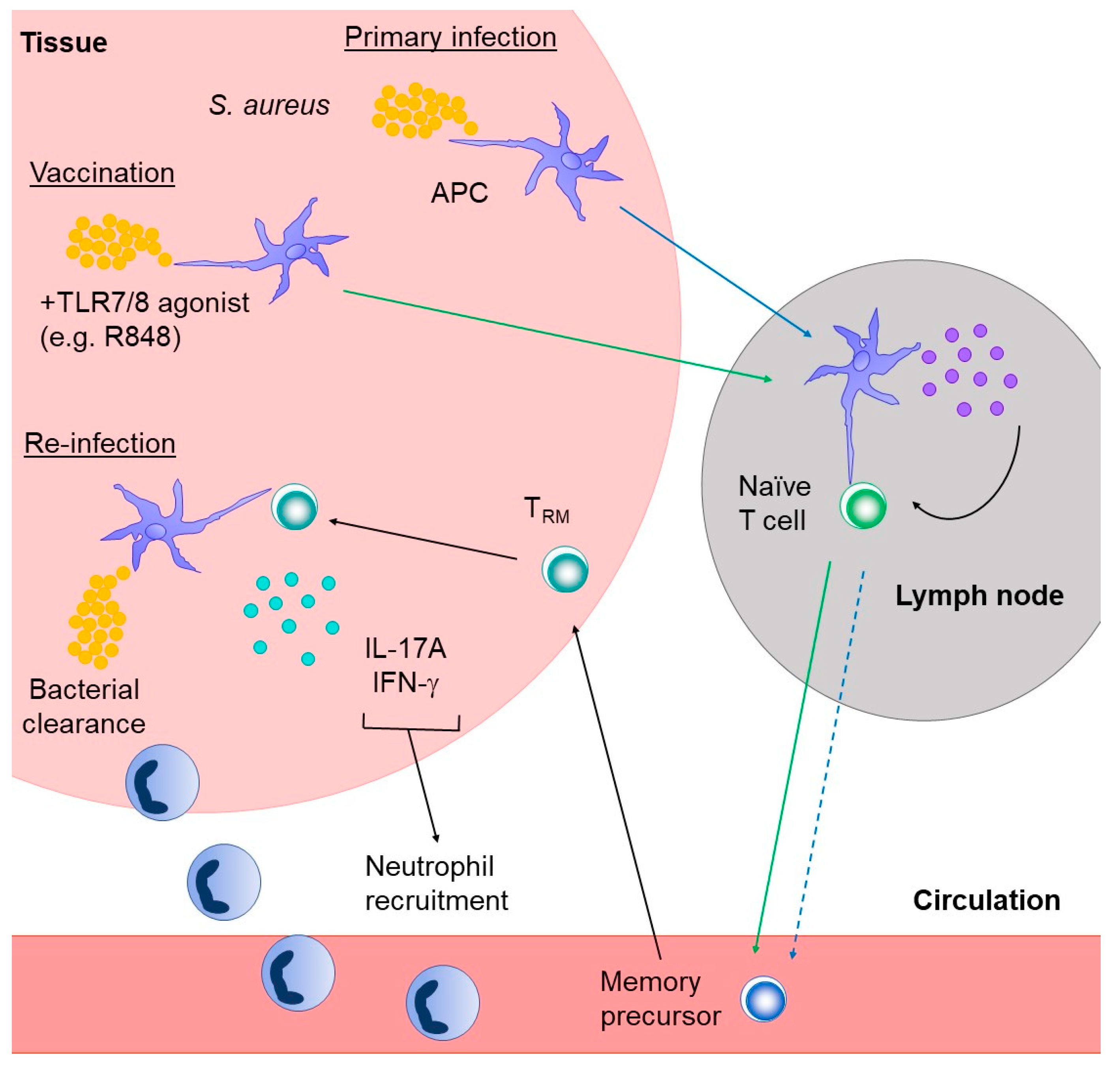
4.2.3. Tissue Resident Memory T Cells
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, D.J.; Sexton, D.J.; Kanafani, Z.A.; Auten, G.; Kaye, K.S. Severe surgical site infection in community hospitals: Epidemiology, key procedures, and the changing prevalence of methicillin-resistant Staphylococcus aureus. Infect. Control. Hosp. Epidemiol. 2007, 28, 1047–1053. [Google Scholar] [CrossRef]
- Berríos-Torres, S.I.; Umscheid, C.A.; Bratzler, D.W.; Leas, B.; Stone, E.C.; Kelz, R.R.; Reinke, C.E.; Morgan, S.; Solomkin, J.S.; Mazuki, J.E.; et al. Centers for Disease Control and Prevention Guideline for the Prevention of Surgical Site Infection. JAMA Surg. 2017, 152, 784–791. [Google Scholar] [CrossRef]
- Peng, H.; Liu, D.; Ma, Y.; Gao, W. Comparison of community- and healthcare-associated methicillin-resistant Staphylococcus aureus isolates at a Chinese tertiary hospital, 2012–2017. Sci. Rep. 2018, 8, 17916. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Singh, A.; David, M.Z.; Bartsch, S.M.; Slayton, R.B.; Huang, S.S.; Zimmer, S.M.; Potter, M.A.; Macal, C.M.; Lauderdale, D.S.; et al. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin. Microbiol. Infect. 2013, 19, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acton, D.S.; Plat-Sinnige, M.J.; van Wamel, W.; de Groot, N.; van Belkum, A. Intestinal carriage of Staphylococcus aureus: How does its frequency compare with that of nasal carriage and what is its clinical impact? Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Belkum, A.; Verkaik, N.J.; de Vogel, C.P.; Boelens, H.A.; Verveer, J.; Nouwen, J.L.; Verbrugh, H.A.; Wertheim, H.F. Reclassification of Staphylococcus aureus nasal carriage types. J. Infect. Dis. 2009, 199, 1820–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Truque, N.; Tedeschi, S.; Saye, E.J.; McKenna, B.D.; Langdon, W.; Wright, J.P.; Alsentzer, A.; Arnold, S.; Saville, B.R.; Wang, W.; et al. Relationship between maternal and neonatal Staphylococcus aureus colonization. Pediatrics 2012, 129, e1252-9. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.G.; Diep, B.A. Clinical practice: Colonization, fomites, and virulence: Rethinking the pathogenesis of community-associated methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008, 46, 752–760. [Google Scholar] [CrossRef]
- Skinner, D.; Keefer, S.C. Significance of bacteremia caused by staphylococcus aureus: A study of one hundred and twenty-two cases and a review of the literature concerned with experimental infection in animals. Arch. Intern. Med. 1941, 68, 851–875. [Google Scholar] [CrossRef]
- van Hal, S.J.; Jensen, S.O.; Vaska, V.L.; Espedido, B.A.; Paterson, D.L.; Gosbell, I.B. Predictors of mortality in Staphylococcus aureus Bacteremia. Clin. Microbiol. Rev. 2012, 25, 362–386. [Google Scholar] [CrossRef] [Green Version]
- Kopp, B.J.; Nix, D.E.; Armstrong, E.P. Clinical and economic analysis of methicillin-susceptible and -resistant Staphylococcus aureus infections. Ann. Pharm. 2004, 38, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Giersing, B.K.; Dastgheyb, S.S.; Modjarrad, K.; Moorthy, V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine 2016, 34, 2962–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redi, D.; Raffaelli, C.S.; Rossetti, B.; De Luca, A.; Montagnani, F. Staphylococcus aureus vaccine preclinical and clinical development: Current state of the art. N. Microbiol. 2018, 41, 208–213. [Google Scholar]
- Fowler, V.G., Jr.; Proctor, R.A. Where does a Staphylococcus aureus vaccine stand? Clin. Microbiol. Infect. 2014, 20 (Suppl. S5), 66–75. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.S.; Miller, A.A.; Donald, R.G.; Scully, I.L.; Nanra, J.S.; Cooper, D.; Jansen, K.U. Development of a multicomponent Staphylococcus aureus vaccine designed to counter multiple bacterial virulence factors. Hum. Vaccines Immunother. 2012, 8, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnoli, F.; Bertholet, S.; Grandi, G. Inferring reasons for the failure of Staphylococcus aureus vaccines in clinical trials. Front. Cell Infect. Microbiol. 2012, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, D.; Grumann, D.; Trübe, P.; Pritchett-Corning, K.; Johnson, S.; Reppschläger, K.; Gumz, J.; Sundaramoorthy, N.; Michalik, S.; Berg, S.; et al. Laboratory Mice Are Frequently Colonized with Staphylococcus aureus and Mount a Systemic Immune Response-Note of Caution for In vivo Infection Experiments. Front. Cell Infect. Microbiol. 2017, 7, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchett-Corning, K.R.; Cosentino, J.; Clifford, C.B. Contemporary prevalence of infectious agents in laboratory mice and rats. Lab. Anim. 2009, 43, 165–173. [Google Scholar] [CrossRef]
- Benjamin, D.K.; Schelonka, R.; White, R.; Holley, H.P.; Bifano, E.; Cummings, J.; Adcock, K.; Kaufman, D.; Puppala, B.; Riedel, P.; et al. A blinded, randomized, multicenter study of an intravenous Staphylococcus aureus immune globulin. J. Perinatol. 2006, 26, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Rupp, M.E.; Holley, H.P., Jr.; Lutz, J.; Dicpinigaitis, P.V.; Woods, C.W.; Levine, D.P.; Veney, N.; Fowler, V.G., Jr. Phase II, randomized, multicenter, double-blind, placebo-controlled trial of a polyclonal anti-Staphylococcus aureus capsular polysaccharide immune globulin in treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2007, 51, 4249–4254. [Google Scholar] [CrossRef] [Green Version]
- DeJonge, M.; Burchfield, D.; Bloom, B.; Duenas, M.; Walker, W.; Polak, M.; Jung, E.; Millard, D.; Schelonka, R.; Eyal, F.; et al. Clinical trial of safety and efficacy of INH-A21 for the prevention of nosocomial staphylococcal bloodstream infection in premature infants. J. Pediatr. 2007, 151, 260–265.e1. [Google Scholar] [CrossRef] [PubMed]
- Weems, J.J., Jr.; Steinberg, J.P.; Filler, S.; Baddley, J.W.; Corey, G.R.; Sampathkumar, P.; Winston, L.; John, J.F.; Kubin, C.J.; Talwani, R.; et al. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2006, 50, 2751–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magyarics, Z.; Leslie, F.; Bartko, J.; Rouha, H.; Luperchio, S.; Schörgenhofer, C.; Schwameis, M.; Derhaschnig, U.; Lagler, H.; Stiebellehner, L.; et al. Randomized, Double-Blind, Placebo-Controlled, Single-Ascending-Dose Study of the Penetration of a Monoclonal Antibody Combination (ASN100) Targeting Staphylococcus aureus Cytotoxins in the Lung Epithelial Lining Fluid of Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63, e00350-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisman, L.E.; Thackray, H.M.; Steinhorn, R.H.; Walsh, W.F.; Lassiter, H.A.; Dhanireddy, R.; Brozanski, B.S.; Palmer, K.G.; Trautman, M.S.; Escobedo, M.; et al. A randomized study of a monoclonal antibody (pagibaximab) to prevent staphylococcal sepsis. Pediatrics 2011, 128, 271–279. [Google Scholar] [CrossRef]
- Yu, X.Q.; Robbie, G.J.; Wu, Y.; Esse, M.T.; Jensen, K.; Schwartz, H.I.; Bellamy, T.; Hernandez-Illas, M.; Jafri, H.S. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, Extended-Half-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemother. 2017, 61, e01020-16. [Google Scholar] [CrossRef] [Green Version]
- François, B.; Mercier, E.; Gonzalez, C.; Asehnoune, K.; Nseir, S.; Fiancette, M.; Desachy, A.; Plantefève, G.; Meziani, F.; de Lame, P.A.; et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: First-in-human trial. Intensive Care Med. 2018, 44, 1787–1796. [Google Scholar] [CrossRef]
- Fowler, V.G.; Allen, K.B.; Moreira, E.D.; Moustafa, M.; Isgro, F.; Boucher, H.W.; Corey, R.; Carmeli, C.; Betts, R.; Harzel, J.S.; et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: A randomized trial. JAMA 2013, 309, 1368–1378. [Google Scholar] [CrossRef]
- McNeely, T.B.; Shah, N.A.; Fridman, A.; Joshi, A.; Hartzel, J.S.; Keshari, R.S.; Lupu, F.; DiNubile, M.J. Mortality among recipients of the Merck V710 Staphylococcus aureus vaccine after postoperative S. aureus infections: An analysis of possible contributing host factors. Hum. Vaccines Immunother. 2014, 10, 3513–3516. [Google Scholar] [CrossRef] [Green Version]
- Aman, M.J. Integrated BioTherapeutics. Hum. Vaccines Immunother. 2018, 14, 1308–1310. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.L.; Lalani, T.; Niknian, M.; Maguire, J.D.; Hospenthal, D.R.; Fattom, A.; Taylor, K.; Fraser, J.; Wilkins, K.; Ellis, M.W.; et al. Safety and immunogenicity of a recombinant Staphylococcus aureus α-toxoid and a recombinant Panton-Valentine leukocidin subunit, in healthy adults. Hum. Vaccines Immunother. 2017, 13, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.H.; Pasetti, M.F.; Adhikari, R.P.; Baughman, H.; Douglas, R.; El-Khorazaty, J.; Greenberg, N.; Holtsberg, F.W.; Liao, G.C.; Reymann, M.K.; et al. Safety and Immunogenicity of a Parenterally Administered, Structure-Based Rationally Modified Recombinant Staphylococcal Enterotoxin B Protein Vaccine, STEBVax. Clin. Vaccines Immunol. 2016, 23, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.S.; White, C.J.; Ibrahim, A.S.; Filler, S.G.; Fu, Y.; Yeaman, M.R.; Edwards, J.E., Jr.; Hennessey, J.P., Jr. NDV-3, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine 2012, 30, 7594–7600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtman, A.; Begier, E.; Mohamed, N.; Baber, J.; Sabharwal, C.; Haupt, R.M.; Edwards, H.; Cooper, D.; Jansen, K.U.; Anderson, A.S. The development of a staphylococcus aureus four antigen vaccine for use prior to elective orthopedic surgery. Hum. Vaccines Immunother. 2019, 15, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Fattom, A.; Matalon, A.; Buerkert, J.; Taylor, K.; Damaso, S.; Boutriau, D. Efficacy profile of a bivalent Staphylococcus aureus glycoconjugated vaccine in adults on hemodialysis: Phase III randomized study. Hum. Vaccines Immunother. 2015, 11, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Fattom, A.I.; Horwith, G.; Fuller, S.; Propst, M.; Naso, R. Development of StaphVAX, a polysaccharide conjugate vaccine against S. aureus infection: From the lab bench to phase III clinical trials. Vaccine 2004, 22, 880–887. [Google Scholar] [CrossRef]
- Spaan, A.N.; Surewaard, B.G.; Nijland, R.; van Strijp, J.A. Neutrophils versus Staphylococcus aureus: A biological tug of war. Annu. Rev. Microbiol. 2013, 67, 629–650. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, I.P.; Liu, G.Y. Targeting fundamental pathways to disrupt Staphylococcus aureus survival: Clinical implications of recent discoveries. JCI Insight 2018, 3, e98216. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, H.S.; Hamburger, J.B.; Ahn, S.H.; McCafferty, D.G.; Yang, S.R.; Fowler, V.G., Jr. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect. Immun. 2009, 77, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.S.; O’Connell, R.M.; Gutierrez, M.A.; Pietras, E.M.; Shahangian, A.; Gross, C.E.; Thirumala, A.; Cheung, A.L.; Cheng, G.; Modlin, R.L. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 2006, 24, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Hoshino, K.; Akira, S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 2000, 165, 5392–5396. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.L.; von Bernuth, H.; Picard, C.; Zhang, S.Y.; Chang, H.H.; Yang, K.; Chrabieh, M.; Issekutz, A.C.; Cunningham, C.K.; Gallin, J.; et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J. Exp. Med. 2007, 204, 2407–2422. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Puel, A.; Bonnet, M.; Ku, C.L.; Bustamante, J.; Yang, K.; Soudais, C.; Dupuis, S.; Feinberg, J.; Fieschi, C.; et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003, 299, 2076–2079. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; von Bernuth, H.; Ghandil, P.; Chrabieh, M.; Levy, O.; Arkwright, P.D.; McDonald, D.; Geha, R.S.; Takada, H.; Krause, J.C.; et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine 2010, 89, 403–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Bernuth, H.; Picard, C.; Jin, Z.; Pankla, R.; Xiao, H.; Ku, C.L.; Chrabieh, M.; Mustapha, I.B.; Ghandil, P.; Camcioglu, Y.; et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 2008, 321, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Olaniyi, R.; Kwiecinski, J.M.; Wardenburg, J.B. Staphylococcus aureus toxin suppresses antigen-specific T cell responses. J. Clin. Investig. 2020, 130, 1122–1127. [Google Scholar] [CrossRef] [Green Version]
- Andrews, T.; Sullivan, K.E. Infections in patients with inherited defects in phagocytic function. Clin. Microbiol. Rev. 2003, 16, 597–621. [Google Scholar] [CrossRef] [Green Version]
- Bouma, G.; Ancliff, P.J.; Thrasher, A.J.; Burns, S.O. Recent advances in the understanding of genetic defects of neutrophil number and function. Br. J. Haematol. 2010, 151, 312–326. [Google Scholar] [CrossRef]
- González-Barca, E.; Carratalà, J.; Mykietiuk, A.; Fernández-Sevilla, A.; Gudiol, F. Predisposing factors and outcome of Staphylococcus aureus bacteremia in neutropenic patients with cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2001, 20, 117–119. [Google Scholar] [CrossRef]
- Lakshman, R.; Finn, A. Neutrophil disorders and their management. J. Clin. Pathol. 2001, 54, 7–19. [Google Scholar] [CrossRef]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; Van Kessel, K.P.; Van Strijp, J.A.; Götz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis. 2002, 186, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Essex, A.; Buchanan, J.T.; Datta, V.; Hoffman, H.M.; Bastian, J.F.; Fierer, J.; Nizet, V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005, 202, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, M.; Kolar, S.L.; Müller, S.; Reyes, C.N.; Wolf, A.J.; Ogawa, C.; Singhania, R.; De Carvalho, D.D.; Arditi, M.; Underhill, D.M.; et al. O-Acetylation of Peptidoglycan Limits Helper T Cell Priming and Permits Staphylococcus aureus Reinfection. Cell Host Microbe 2017, 22, 543–551.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero Pastrana, F.; Neef, J.; Koedijk, D.; de Graaf, D.; Duipmans, J.; Jonkman, M.F.; Engelmann, S.; van Dijl, J.M.; Buist, G. Human antibody responses against non-covalently cell wall-bound Staphylococcus aureus proteins. Sci. Rep. 2018, 8, 3234. [Google Scholar] [CrossRef]
- Stolz, S.J.; Davis, J.P.; Vergeront, J.M.; Crass, B.A.; Chesney, P.J.; Wand, P.J.; Bergdoll, M.S. Development of serum antibody to toxic shock toxin among individuals with toxic shock syndrome in Wisconsin. J. Infect. Dis. 1985, 151, 883–889. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, X.; Akhgar, A.; Li, J.J.; Mok, H.; Sellman, B.R.; Yu, L.; Roskos, L.K.; Esser, M.T.; Ruzin, A. Prevalence of IgG and Neutralizing Antibodies against Staphylococcus aureus Alpha-Toxin in Healthy Human Subjects and Diverse Patient Populations. Infect. Immun. 2018, 86, e00671-17. [Google Scholar]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Goodyear, C.S.; Silverman, G.J. Death by a B cell superantigen: In vivo VH-targeted apoptotic supraclonal B cell deletion by a Staphylococcal Toxin. J. Exp. Med. 2003, 197, 1125–1139. [Google Scholar] [CrossRef]
- Adhikari, R.P.; Ajao, A.O.; Aman, M.J.; Karauzum, H.; Sarwar, J.; Lydecker, A.D.; Johnson, J.K.; Nguyen, C.; Chen, W.H.; Roghmann, M.C. Lower antibody levels to Staphylococcus aureus exotoxins are associated with sepsis in hospitalized adults with invasive S. aureus infections. J. Infect. Dis. 2012, 206, 915–923. [Google Scholar] [CrossRef]
- Fritz, S.A.; Tiemann, K.M.; Hogan, P.G.; Epplin, E.K.; Rodriguez, M.; Al-Zubeidi, D.N.; Bubeck Wardenburg, J.; Hunstad, D.A. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin. Infect. Dis. 2013, 56, 1554–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.M.; Soper, N.; Bennett, M.; Fallon, J.K.; Michell, A.R.; Alter, G.; Liu, G.Y.; Thomsen, I. Adoptive Transfer of Serum Samples from Children with Invasive Staphylococcal Infection and Protection Against Staphylococcus aureus Sepsis. J. Infect. Dis. 2020, jiaa482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ledue, O.; Jun, M.; Goulart, C.; Malley, R.; Lu, Y.J. Protection against Staphylococcus aureus Colonization and Infection by B- and T-Cell-Mediated Mechanisms. mBio 2018, 9, e01949-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, M.J.; Caldera, J.R.; Nguyen, C.; Sharma, P.; Castro, A.M.; Kolar, S.L.; Tsai, C.M.; Limon, J.J.; Becker, C.A.; Martins, G.A.; et al. Harnessing antifungal immunity in pursuit of a Staphylococcus aureus vaccine strategy. PLoS Pathog. 2020, 16, e1008733. [Google Scholar] [CrossRef]
- Dhalla, F.; Misbah, S.A. Secondary antibody deficiencies. Curr. Opin Allergy Clin. Immunol. 2015, 15, 505–513. [Google Scholar] [CrossRef]
- Hoernes, M.; Seger, R.; Reichenbach, J. Modern management of primary B-cell immunodeficiencies. Pediatr. Allergy Immunol. 2011, 22, 758–769. [Google Scholar] [CrossRef]
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Investif. 2010, 120, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Marchitto, M.C.; Dillen, C.A.; Liu, H.; Miller, R.J.; Archer, N.K.; Ortines, R.V.; Alphonse, M.P.; Marusina, A.I.; Merleev, A.A.; Wang, Y.; et al. Clonal Vγ6(+)Vδ4(+) T cells promote IL-17-mediated immunity against Staphylococcus aureus skin infection. Proc. Natl. Acad. Sci. USA 2019, 116, 10917–10926. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, B.S.; Romagnoli, P.A.; Pham, Q.M.; Fu, H.H.; Alonzo, F., 3rd; Schubert, W.D.; Freitag, N.E.; Lefrançois, L. γδ T cells exhibit multifunctional and protective memory in intestinal tissues. Immunity 2013, 39, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4⁺T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [Green Version]
- Lowy, F.D. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol. 2000, 8, 341–343. [Google Scholar] [CrossRef]
- Joshi, A.; Pancari, G.; Cope, L.; Bowman, E.P.; Cua, D.; Proctor, R.A.; McNeely, T. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum. Vaccines Immunother. 2012, 8, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, C.P.; Daniels, M.; Zhao, F.; Alegre, M.L.; Chong, A.S.; Daum, R.S. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infect. Immun. 2014, 82, 2125–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.Y.; Leech, J.M.; Rogers, T.R.; McLoughlin, R.M. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J. Clin. Investig. 2002, 110, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., 3rd; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crum-Cianflone, N.F.; Grandits, G.; Echols, S.; Ganesan, A.; Landrum, M.; Weintrob, A.; Barthel, R.; Agan, B.; Infectious Disease Clinical Research Program. Trends and causes of hospitalizations among HIV-infected persons during the late HAART era: What is the impact of CD4 counts and HAART use? J. Acquir Immun. Defic. Syndr. 2010, 54, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, R.; Calza, L.; Chiodo, F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: A 10-year survey. J. Cutan. Pathol. 2002, 29, 168–172. [Google Scholar] [CrossRef]
- Manfredi, R.; Costigliola, P.; Ricchi, E.; Chiodo, F. Sepsis-bacteraemia and other infections due to non-opportunistic bacterial pathogens in a consecutive series of 788 patients hospitalized for HIV infection. Clin. Ter. 1993, 143, 279–290. [Google Scholar]
- Beekhuizen, H.; van de Gevel, J.S. Gamma Interferon Confers Resistance to Infection with Staphylococcus aureus in Human Vascular Endothelial Cells by Cooperative Proinflammatory and Enhanced Intrinsic Antibacterial Activities. Infect. Immun. 2007, 75, 5615–5626. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.R.; Lalor, S.J.; McLoughlin, R.M. Activation of Human Vδ2+ γδ T Cells by Staphylococcus aureus Promotes Enhanced Anti-Staphylococcal Adaptive Immunity. J. Immunol. 2020, 205, 1039–1049. [Google Scholar] [CrossRef]
- De Forge, L.E.; Billeci, K.L.; Kramer, S.M. Effect of IFN-gamma on the killing of S. aureus in human whole blood. Assessment of bacterial viability by CFU determination and by a new method using alamarBlue. J. Immunol. Methods 2000, 245, 79–89. [Google Scholar]
- Brown, A.F.; Murphy, A.G.; Lalor, S.J.; Leech, J.M.; O’Keeffe, K.M.; Mac Aogáin, M.; O’Halloran, D.P.; Lacey, K.A.; Tavakol, M.; Hearnden, C.H.; et al. Memory Th1 Cells Are Protective in Invasive Staphylococcus aureus Infection. PLoS Pathog. 2015, 11, e1005226. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Hu, D.L.; Mori, F.; Wakabayashi, K.; Iwakura, Y.; Nakane, A. Role of interleukin-17A in cell-mediated protection against Staphylococcus aureus infection in mice immunized with the fibrinogen-binding domain of clumping factor A. Infect. Immun. 2010, 78, 4234–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Ibrahim, A.S.; Xu, X.; Farber, J.M.; Avanesian, V.; Baquir, B.; Fu, Y.; French, S.W.; Edwards, J.E., Jr.; Spellberg, B. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 2009, 5, e1000703. [Google Scholar] [CrossRef]
- Nurjadi, D.; Kain, M.; Marcinek, P.; Gaile, M.; Heeg, K.; Zanger, P. Ratio of T-Helper Type 1 (Th1) to Th17 Cytokines in Whole Blood Is Associated with Human β-Defensin 3 Expression in Skin and Persistent Staphylococcus aureus Nasal Carriage. J. Infect. Dis. 2016, 214, 1744–1751. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [Green Version]
- Renner, E.D.; Rylaarsdam, S.; Anover-Sombke, S.; Rack, A.L.; Reichenbach, J.; Carey, J.C.; Zhu, Q.; Jansson, A.F.; Barboza, J.; Schimke, L.F.; et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2008, 122, 181–187. [Google Scholar] [CrossRef]
- Wu, J.; Ding, Y.; Wang, J.; Wang, F. Staphylococcus aureus induces TGF-β(1) and bFGF expression through the activation of AP-1 and NF-κB transcription factors in bovine mammary epithelial cells. Microb. Pathog. 2018, 117, 276–284. [Google Scholar] [CrossRef]
- Zhang, L.J.; Chen, S.X.; Guerrero-Juarez, C.F.; Li, F.; Tong, Y.; Liang, Y.; Liggins, M.; Chen, X.; Chen, H.; Li, M.; et al. Age-Related Loss of Innate Immune Antimicrobial Function of Dermal Fat Is Mediated by Transforming Growth Factor Beta. Immunity 2019, 50, 121–136.e5. [Google Scholar] [CrossRef] [Green Version]
- Leech, J.M.; Lacey, K.A.; Mulcahy, M.E.; Medina, E.; McLoughlin, R.M. IL-10 Plays Opposing Roles during Staphylococcus aureus Systemic and Localized Infections. J. Immunol. 2017, 198, 2352–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjertsson, I.; Hultgren, O.H.; Tarkowski, A. Interleukin-10 ameliorates the outcome of Staphylococcus aureus arthritis by promoting bacterial clearance. Clin. Exp. Immunol. 2002, 130, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.E.; Shukla, S.K.; Berti, A.D.; Hayney, M.S.; Henriquez, K.M.; Ranzoni, A.; Cooper, M.A.; Proctor, R.A.; Nizet, V.; et al. Increased Endovascular Staphylococcus aureus Inoculum Is the Link Between Elevated Serum Interleukin 10 Concentrations and Mortality in Patients With Bacteremia. Clin. Infect. Dis. 2017, 64, 1406–1412. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrançois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Bankovich, A.J.; Shiow, L.R.; Cyster, J.G. CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J. Biol. Chem. 2010, 285, 22328–22337. [Google Scholar] [CrossRef] [Green Version]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.I.; Brolin, R.E.; Ebert, E.C. Integrin alpha1beta1 (VLA-1) mediates adhesion of activated intraepithelial lymphocytes to collagen. Immunology 1999, 97, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.J.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 2017, 552, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Wynne-Jones, E.; Freestone, D.; Pellicci, D.G.; Mielke, L.A.; Newman, D.M.; Braun, A.; Masson, F.; Kallies, A.; Belz, G.T.; et al. T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 2015, 43, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Nath, A.P.; Braun, A.; Ritchie, S.C.; Carbone, F.R.; Mackay, L.K.; Gebhardt, T.; Inouye, M. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 2019, 14, e0210495. [Google Scholar] [CrossRef]
- Mani, V.; Bromley, S.K.; Äijö, T.; Mora-Buch, R.; Carrizosa, E.; Warner, R.D.; Hamze, M.; Sen, D.R.; Chasse, A.Y.; Lorant, A.; et al. Migratory DCs activate TGF-β to precondition naïve CD8+ T cells for tissue-resident memory fate. Science 2019, 366, eaav5728. [Google Scholar] [CrossRef]
- Thompson, E.A.; Thompson, E.A.; Darrah, P.A.; Foulds, K.E.; Hoffer, E.; Caffrey-Carr, A.; Norenstedt, S.; Perbeck, L.; Seder, R.A.; Kedl, R.M.; et al. Monocytes Acquire the Ability to Prime Tissue-Resident T Cells via IL-10-Mediated TGF-β Release. Cell Rep. 2019, 28, 1127–1135.e4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Bevan, M.J. Transforming Growth Factor-β Signaling Controls the Formation and Maintenance of Gut-Resident Memory T Cells by Regulating Migration and Retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Wilk, M.M.; Misiak, A.; McManus, R.M.; Allen, A.C.; Lynch, M.A.; Mills, K. Lung CD4 Tissue-Resident Memory T Cells Mediate Adaptive Immunity Induced by Previous Infection of Mice with Bordetella pertussis. J. Immunol. 2017, 199, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Iwanaga, N.; Sandquist, I.; Chen, K.; Norton, E.B.; Rangel-Moreno, J.; Kolls, J.K. Klebsiella Pneumoniae Mucosal Vaccination Elicits Lung CD4+ TRM cells that are resistant to CD4 depleting antibodies. J. Immunol. 2020, 204 (Suppl. S1), 217–234. [Google Scholar]
- Stary, G.; Olive, A.; Radovic-Moreno, A.F.; Gondek, D.; Alvarez, D.; Basto, P.A.; Perro, M.; Vrbanac, V.D.; Tager, A.M.; Shi, J.; et al. VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 2015, 348, aaa8205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zens, K.D.; Chen, J.K.; Farber, D.L. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2016, 1, e85832. [Google Scholar] [CrossRef] [PubMed]
- Piet, B.; de Bree, G.J.; Smids-Dierdorp, B.S.; van der Loos, C.M.; Remmerswaal, E.B.; von der Thüsen, J.H.; van Haarst, J.M.; Eerenberg, J.P.; ten Brinke, A.; van der Bij, W.; et al. CD8⁺ T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Investig. 2011, 121, 2254–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzolla, A.; Nguyen, T.H.; Sant, S.; Jaffar, J.; Loudovaris, T.; Mannering, S.I.; Thomas, P.G.; Westall, G.P.; Kedzierska, K.; Wakim, L.M. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J. Clin. Investig. 2018, 128, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Posavad, C.M.; Zhao, L.; Dong, L.; Jin, L.; Stevens, C.E.; Magaret, A.S.; Johnston, C.; Wald, A.; Zhu, J.; Corey, L.; et al. Enrichment of herpes simplex virus type 2 (HSV-2) reactive mucosal T cells in the human female genital tract. Mucosal. Immunol. 2017, 10, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.L.; Miron, M.; Thome, J.J.; Matsuoka, N.; Weiner, J.; Rak, M.A.; Igarashi, S.; Granot, T.; Lerner, H.; Goodrum, F.; et al. Tissue reservoirs of antiviral T cell immunity in persistent human CMV infection. J. Exp. Med. 2017, 214, 651–667. [Google Scholar] [CrossRef]
- Jozwik, A.; Habibi, M.S.; Paras, A.; Zhu, J.; Guvenel, A.; Dhariwal, J.; Almond, M.; Wong, E.; Sykes, A.; et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat. Commun. 2015, 6, 10224. [Google Scholar] [CrossRef]
- Glennie, N.D.; Yeramilli, V.A.; Beiting, D.P.; Volk, S.W.; Weaver, C.T.; Scott, P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J. Exp. Med. 2015, 212, 1405–1414. [Google Scholar] [CrossRef]
- Hobbs, S.J.; Osborn, J.F.; Nolz, J.C. Activation and trafficking of CD8(+) T cells during viral skin infection: Immunological lessons learned from vaccinia virus. Curr. Opin. Virol. 2018, 28, 12–19. [Google Scholar] [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, P.; Swan, D.A.; Duke, E.; Corey, L.; Zhu, J.; Davé, V.; Spuhler, L.R.; Lund, J.M.; Prlic, M.; Schiffer, J.T. Tissue-resident T cell-derived cytokines eliminate herpes simplex virus-2-infected cells. J. Clin. Investig. 2020, 130, 2903–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linehan, J.L.; Harrison, O.J.; Han, S.J.; Byrd, A.L.; Vujkovic-Cvijin, I.; Villarino, A.V.; Sen, S.K.; Shaik, J.; Smelkinson, M.; Tamoutounour, S.; et al. Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 2018, 172, 784–796.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Antigen | Company/ Name | Phase | Target Group | Result | Study Dates | Notes | Citation |
|---|---|---|---|---|---|---|---|
| CP5/CP8 | Nabi/ Altastaph | II | Treatment of Bacteriemia | Completed | 2002–2004 | Shorter length of stay for treatment group. No further development. | Rupp et al., 2007 [20] |
| CP5/CP8 | Nabi/ Altastaph | II | Prevention of invasive infection in very low birth weight neonates | Failed | 2003–2004 | No difference between placebo and treatment groups. | Benjamin et al., 2006 [19] |
| ClfA | Bristol-Myers Squibb/ Aurexis Tefibazumab | II | Treatment of bacteremia | Failed | 2005 | No differences between groups. | Weems et al., 2006 [22] |
| ATP-binding cassette (ABC) transporter GrfA | Neu Tec Pharma/Aurograb | II | Treatment of deep-seated infection | Failed | 2004–2006 | No difference between groups | Fowler and Proctor 2015 [14] |
| Hla | AstraZeneca/ Suvratoxumab/ MEDI4893 | II | Mechanically ventilated adults | Failed | 2014–2018 | No significant differences between groups. | Yu et al., 2017 [25] |
| Hla, PVL, LukED, g-hemolysin AB and CB (HlgAB, HlgCB) | Arsanis Inc/ ASN100 | II | Mechanically ventilated subjects | Failed | 2016–2018 | Did not achieve endpoint. Ended in futility. | Magyarics et al., 2019 [23] |
| ClfA (S. aureus) and SdrG (S. epidermidis) | Bristol–Myers Squibb/ Veronate/ Inhibitex | III | Sepsis in premature infants | Failed | 2004–2006 | No difference between placebo and treated group. | DeJonge et al., 2007 [21] |
| LTA | Biosynexus incorporate/ Pagibmaximab | II | Prevention of Sepsis in very low birth weight neonates | Failed | 2009–2011 | No significant difference between groups. | Weisman et al., 2011 [24] |
| Hla | Aridis Pharmaceuticals/ AR-301 | III | Treatment of bacterial pneumonia or ventilator-associated pneumonia | Completed | 2019–2020 | Promising phase II trials (Francois et al., 2018) [26] | unpublished |
| Antigen | Company/ Name | Phase | Target Group | Result | Study Dates | Notes | Citation |
|---|---|---|---|---|---|---|---|
| Enterotoxin A and C1, TSST, alpha-hemolysin, LukS, LukF | BioTherapeutics/ IBT-V02 | Pre-clinical | No data | NA | Scheduled to enter phase I clinical trials in 2020 | Aman 2018 [29] | |
| rAT/rLukS-PV | Nabi/ N.A. | I | Completed | 2009–2011 | No further data | Landrum et al., 2017 [30] | |
| Recombinant Enterotoxin B (rSEB) | BioTherapeutics/ STEBVax | I | Completed | 2011–2015 | No further data | Chen et al., 2016 [31] | |
| Unspecified Recombinant protein—bioconjugated—adjuvanted | GSK/ GSK3878858A | I | Ongoing | 2020–2020 | No data | Unpublished | |
| Als3p | NovaDigm Therapeutics/ NDV-3A | II | Nasal colonization (and SSTI) in military personnel | Completed | 2018–2019 | Well tolerated in phase I trials and provided protection against S. aureus and C. albicans | Schmidt et al., 2012 [32] |
| CflA/MntC/CP5/CP8 | Pfizer/ SA-4Ag | IIb/III | Elective orthopedic surgery | Failed | 2015–2019 | Stopped after sub-par results | Gurtman et al., 2019 [33] |
| CP5/CP8 | Nabi/ StaphVax | III | Renal disease or orthopedic surgery | Failed | 2005–2006 | No difference between placebo and vaccinated groups | Fattom et al., 2004; Fattom et al., 2015 [34,35] |
| IsdB | Merck/ V710 | III | Prevention of infection after cardiothoracic surgery | Failed | 2007–2011 | Adverse outcomes in vaccinated group | Fowler et al., 2013; McNeely et al., 2014 [26,27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armentrout, E.I.; Liu, G.Y.; Martins, G.A. T Cell Immunity and the Quest for Protective Vaccines against Staphylococcus aureus Infection. Microorganisms 2020, 8, 1936. https://doi.org/10.3390/microorganisms8121936
Armentrout EI, Liu GY, Martins GA. T Cell Immunity and the Quest for Protective Vaccines against Staphylococcus aureus Infection. Microorganisms. 2020; 8(12):1936. https://doi.org/10.3390/microorganisms8121936
Chicago/Turabian StyleArmentrout, Erin I., George Y. Liu, and Gislâine A. Martins. 2020. "T Cell Immunity and the Quest for Protective Vaccines against Staphylococcus aureus Infection" Microorganisms 8, no. 12: 1936. https://doi.org/10.3390/microorganisms8121936