Identification and Characterization of 33 Bacillus cereus sensu lato Isolates from Agricultural Fields from Eleven Widely Distributed Countries by Whole Genome Sequencing

 , , , , ,
, , , , ,  , , , ,
, , , , 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
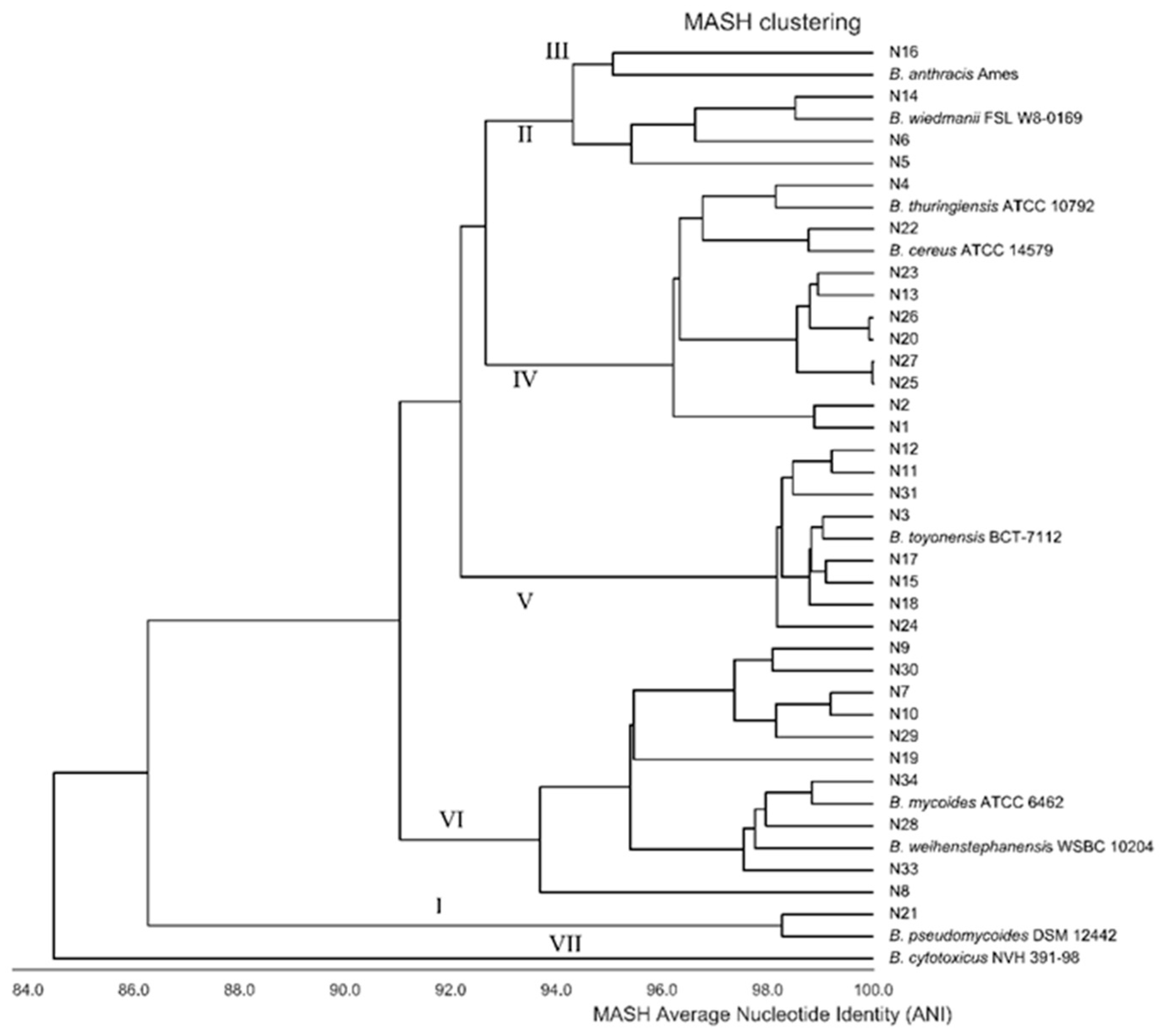
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Priest, F.G.; Barker, M.; Baillie, L.W.J.; Holmes, E.C.; Maiden, M.C.J. Population structure and evolution of the Bacillus cereus group. J. Bacteriol. 2004, 186, 7959–7970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinebretiere, M.H.; Thompson, F.L.; Sorokin, A.; Normand, P.; Dawyndt, P.; Ehling-Schulz, M.; De Vos, P. Ecological diversification in the Bacillus cereus Group. Environ. Microbiol. 2008, 10, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Okinaka, R.T.; Keim, P. The Phylogeny of Bacillus cereus sensu lato. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lai, Q.L.; Goker, M.; Meier-Kolthoff, J.P.; Wang, M.; Sun, Y.M.; Shao, Z.Z. Genomic insights into the taxonomic status of the Bacillus cereus group. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Goker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Bazinet, A.L. Pan-genome and phylogeny of Bacillus cereus sensu lato. BMC Evol. Biol. 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Carroll, L.M.; Wiedmann, M.; Kovac, J. Proposal of a Taxonomic Nomenclature for the Bacillus cereus Group Which Reconciles Genomic Definitions of Bacterial Species with Clinical and Industrial Phenotypes. Mbio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Fiedoruk, K.; Drewnowska, J.M.; Daniluk, T.; Leszczynska, K.; Iwaniuk, P.; Swiecicka, I. Ribosomal background of the Bacillus cereus group thermotypes. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Helgason, E.; Tourasse, N.J.; Meisal, R.; Caugant, D.A.; Kolsto, A.B. Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl. Environ. Microbiol. 2004, 70, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Drewnowska, J.M.; Swiecicka, I. Eco-Genetic Structure of Bacillus cereus sensu lato Populations from Different Environments in Northeastern Poland. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Swiecicka, I.; Bartoszewicz, M.; Kasulyte-Creasey, D.; Drewnowska, J.M.; Murawska, E.; Yernazarova, A.; Mahillon, J. Diversity of thermal ecotypes and potential pathotypes of Bacillus thuringiensis soil isolates. FEMS. Microbiol. Ecol. 2013, 85, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinebretiere, M.H.; Velge, P.; Couvert, O.; Carlin, F.; Debuyser, M.L.; Nguyen-The, C. Ability of Bacillus cereus Group Strains To Cause Food Poisoning Varies According to Phylogenetic Affiliation (Groups I to VII) Rather than Species Affiliation. J. Clin. Microbiol. 2010, 48, 3388–3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottone, E.J. Bacillus cereus, a Volatile Human Pathogen. Clin. Microbiol. Rev. 2010, 23, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehling-Schulz, M.; Lereclus, D.; Koehler, T.M. The Bacillus cereus Group: Bacillus Species with Pathogenic Potential. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Carroll, L.M.; Kovac, J.; Miller, R.A.; Wiedmann, M. Rapid, High-Throughput Identification of Anthrax-Causing and Emetic Bacillus cereus Group Genome Assemblies via BTyper, a Computational Tool for Virulence-Based Classification of Bacillus cereus Group Isolates by Using Nucleotide Sequencing Data. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Travers, R.S.; Martin, P.A.W.; Reichelderfer, C.F. Selective Process for Efficient Isolation of Soil Bacillus spp. Appl. Environ. Microbiol. 1987, 53, 1263–1266. [Google Scholar] [CrossRef] [Green Version]
- Hansen, B.M.; Leser, T.D.; Hendriksen, N.B. Polymerase chain reaction assay for the detection of Bacillus cereus group cells. Fems Microbiol. Lett. 2001, 202, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Willumsen, P.A.; Johansen, J.E.; Karlson, U.; Hansen, B.M. Isolation and taxonomic affiliation of N-heterocyclic aromatic hydrocarbon-transforming bacteria. Appl. Microbiol. Biotechnol. 2005, 67, 420–428. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Lund, O. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, S.; Mayr, R.; Francis, K.P.; Pruss, B.M.; Kaplan, T.; Wiessner-Gunkel, E.; Scherer, S. Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int. J. Syst. Bacteriol. 1998, 48, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.M.; Hoiby, P.E.; Jensen, G.B.; Hendriksen, N.B. The Bacillus cereus bceT enterotoxin sequence reappraised. FEMS Microbiol. Lett. 2003, 223, 21–24. [Google Scholar] [CrossRef] [Green Version]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/bacillus%20cereus (accessed on 5 November 2020).
- Klassen, J.L.; Currie, C.R. Gene fragmentation in bacterial draft genomes: Extent, consequences and mitigation. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, M.J.; Koser, C.U.; Ross, N.E.; Archer, J.A.C. Read Length and Repeat Resolution: Exploring Prokaryote Genomes Using Next-Generation Sequencing Technologies. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Ceuppens, S.; Boon, N.; Uyttendaele, M. Diversity of Bacillus cereus group strains is reflected in their broad range of pathogenicity and diverse ecological lifestyles. FEMS Microbiol. Ecol. 2013, 84, 433–450. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lai, Q.L.; Shao, Z.Z. Genome analysis-based reclassification of Bacillus weihenstephanensis as a later heterotypic synonym of Bacillus mycoides. Int. J. Syst. Evol. Microbiol. 2018, 68, 106–112. [Google Scholar] [CrossRef]
- Bavykin, S.G.; Lysov, Y.P.; Zakhariev, V.; Kelly, J.J.; Jackman, J.; Stahl, D.A.; Cherni, A. Use of 16S rRNA, 23S rRNA, and gyrB gene sequence analysis to determine phylogenetic relationships of Bacillus cereus group microorganisms (vol 42, pg 3711, 2004). J. Clin. Microbiol. 2006, 44, 2676. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beecher, D.J.; Wong, A.C.L. Tripartite haemolysin BL: Isolation and characterization of two distinct homologous sets of components from a single Bacillus cereus isolate. Microbiology-Sgm 2000, 146, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerlund, A.; Ween, A.; Lund, T.; Hardy, S.P.; Granum, P.E. Genetic and functional analysis of the cytK family of genes in Bacillus cereus. Microbiology-Sgm 2004, 150, 2689–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, S.P.; Lund, T.; Granum, P.E. CytK toxin of Bacillus cereus forms pores in planar lipid bilayers and is cytotoxic to intestinal epithelia. FEMS Microbiol. Lett. 2001, 197, 47–51. [Google Scholar] [CrossRef]
- Lund, T.; De Buyser, M.L.; Granum, P.E. A new cytotoxin from Bacillus cereus that may cause necrotic enteritis. Mol. Microbiol. 2000, 38, 254–261. [Google Scholar] [CrossRef]
- von Stetten, F.; Mayr, R.; Scherer, S. Climatic influence on mesophilic Bacillus cereus and psychrotolerant Bacillus weihenstephanensis populations in tropical, temperate and alpine soil. Environ. Microbiol. 1999, 1, 503–515. [Google Scholar] [CrossRef]




{kind=link}
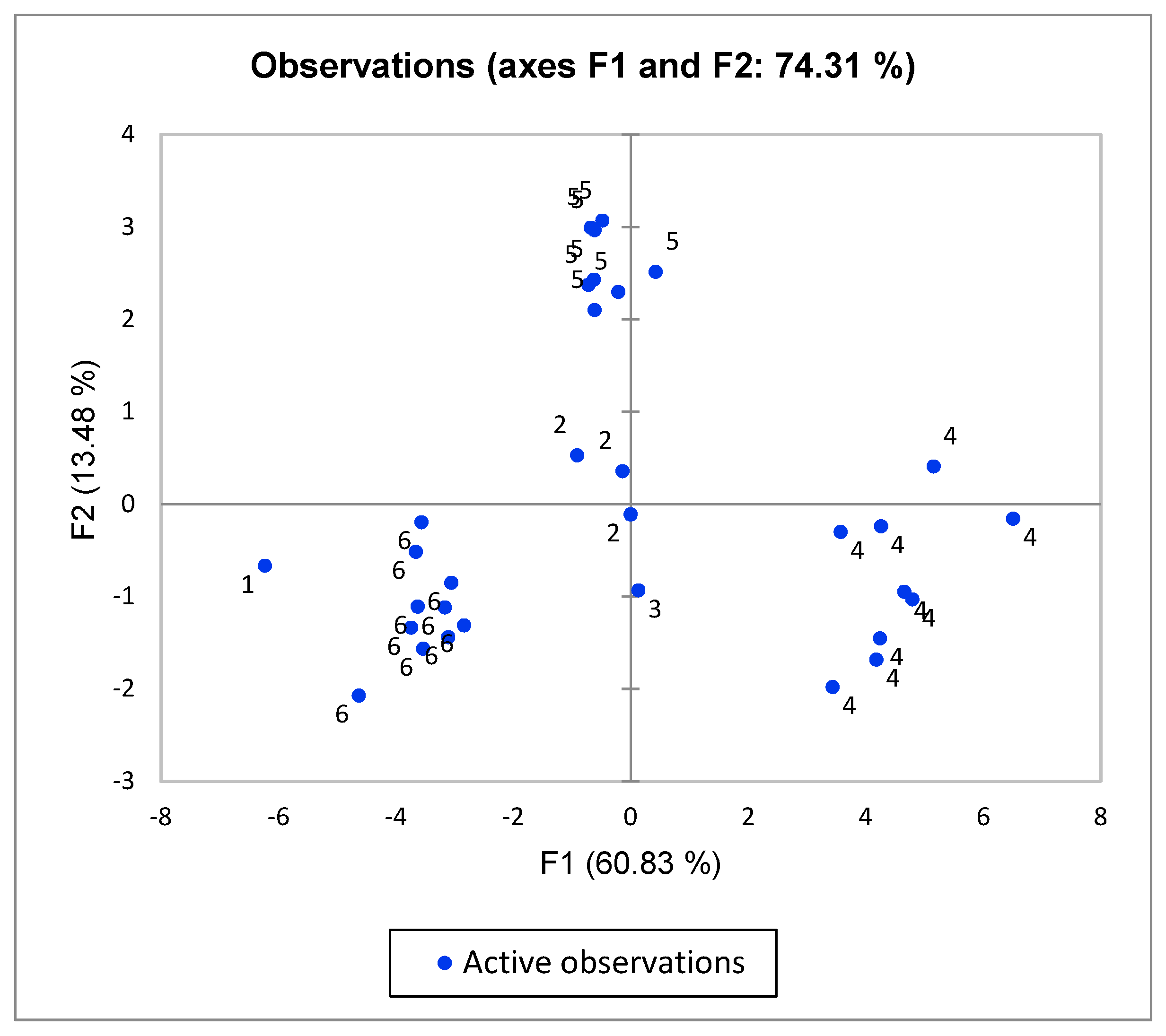{kind=link}
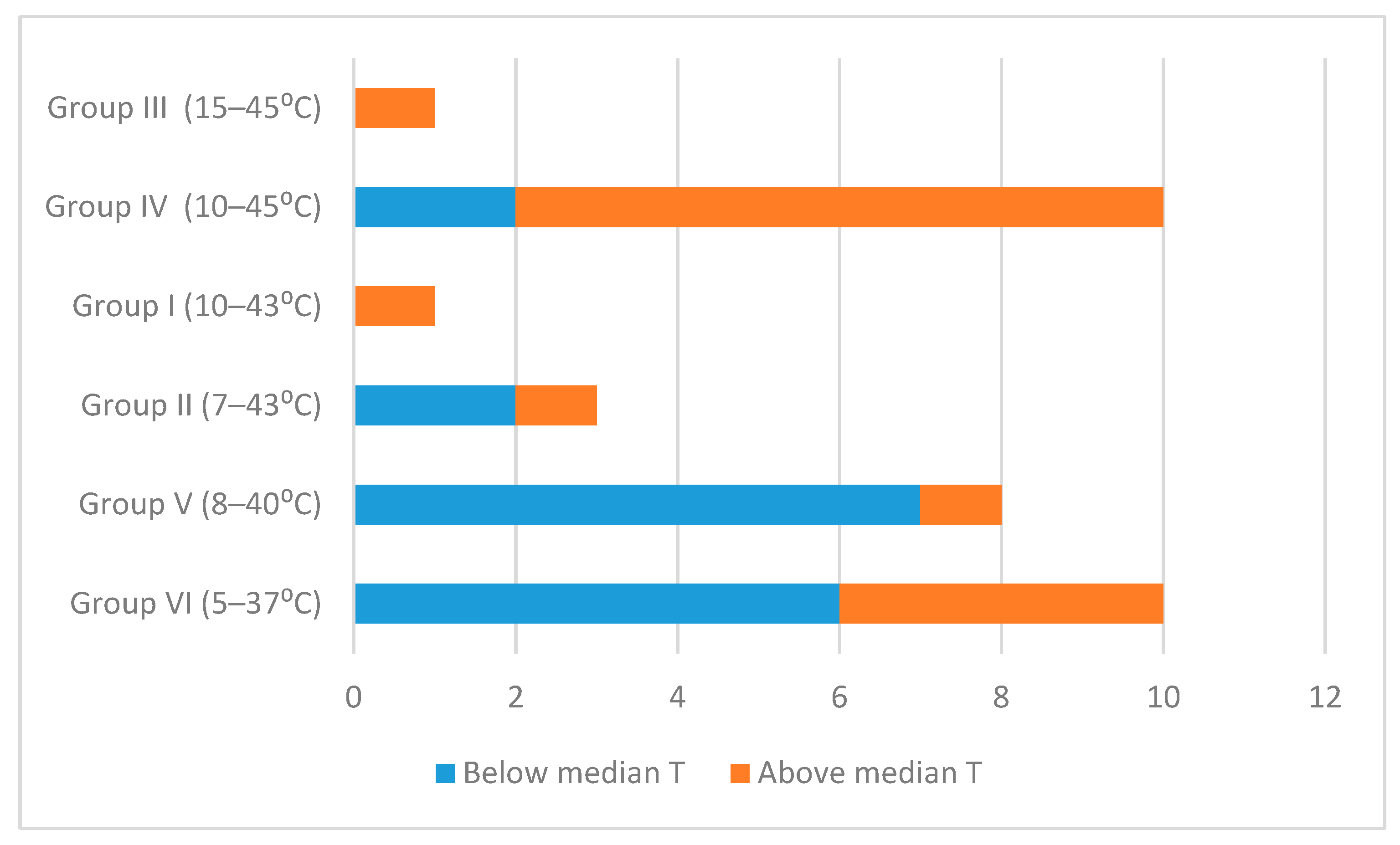{kind=link}
| Isolate Number | Country | GPS-Coordinates | Meters above Sea Level | Crop | Yearly Mean Temperature (°C) |
|---|---|---|---|---|---|
| N1 and N2 | Lithuania | 54.864 23.941 | 50 | Vegetables | 6.4 |
| N3 and N4 | Tunisia | 34.5117 10.4924 | 200 | Olive trees | 18.1 |
| N5 and N6 | Netherlands | 51.966 5.655 | 9 | Leak | 9.2 |
| N7 and N8 | Slovenia | 46.1434 14.5580 | 265 | Oat | 10.4 |
| N9 and N10 | Switzerland | 46.3919 6.21164 | 450 | Livestock | 9.2 |
| N11 and N12 | Hungary | 47.3827 17.4992 | 135 | Maize | 11.1 |
| N13 and N14 | Hungary | 46.0559 19.2622 | 95 | Pea | 11.1 |
| N15 and N16 | Serbia | 45.1908 19.2101 | 88 | Fallow | 11.3 |
| N17 and N18 | Serbia | 45.1908 19.2101 | 88 | Fallow | 11.3 |
| N19 and N20 | Turkey | 39.9672 32.6623 | 938 | Lemon | 10.9 |
| N21 and N22 | Turkey | 40.0808 34.1200 | 780 | Wheat, Barley | 10.9 |
| N23 and N24 | Turkey | 39.9040 32.6345 | 900 | Garden | 10.9 |
| N25 and N26 | Turkey | 39.5456 33.4436 | 1000 | Grass | 10.9 |
| N27 and N28 | Turkey | 40.0047 32.5142 | 1066 | Grass | 10.9 |
| N29 and N30 | Denmark | 55.697 12.103 | 5 | Grass | 7.8 |
| N31 and N32 | Germany | 52.359 13.308 | 42 | Potato | 10.2 |
| N33 and N34 | Belgium | 51.138 3.938 | 6 | Tree Nursery | 10.2 |
| Isolate | Bases | Contigs | CDS | tRNA | rRNA | GC content |
|---|---|---|---|---|---|---|
| N1 | 6589056 | 1486 | 6332 | 139 | 12 | 34.87 |
| N2 | 6208593 | 362 | 6112 | 116 | 10 | 34.7 |
| N3 | 5981260 | 1394 | 5801 | 98 | 10 | 35.18 |
| N4 | 5628374 | 283 | 5577 | 115 | 11 | 35.01 |
| N5 | 6039339 | 435 | 6023 | 120 | 9 | 35.08 |
| N6 | 6050091 | 858 | 6102 | 139 | 12 | 35.33 |
| N7 | 7090662 | 4682 | 6057 | 129 | 14 | 37.86 |
| N8 | 5963382 | 392 | 6054 | 102 | 12 | 35.2 |
| N9 | 5731016 | 563 | 5730 | 112 | 12 | 35.31 |
| N10 | 5648760 | 410 | 5647 | 128 | 11 | 35.25 |
| N11 | 6015328 | 286 | 5869 | 109 | 12 | 35.06 |
| N12 | 6179004 | 1900 | 5902 | 125 | 16 | 35.4 |
| N13 | 5920438 | 304 | 5853 | 112 | 13 | 34.9 |
| N14 | 5687870 | 332 | 5733 | 132 | 8 | 35.17 |
| N15 | 6040251 | 455 | 5965 | 126 | 12 | 34.99 |
| N16 | 5783502 | 405 | 5769 | 120 | 11 | 35.09 |
| N17 | 6225349 | 391 | 6198 | 98 | 7 | 34.91 |
| N18 | 5976955 | 349 | 5862 | 94 | 10 | 35.05 |
| N19 | 6310391 | 1305 | 6317 | 105 | 9 | 35.3 |
| N20 | 6366475 | 989 | 6433 | 104 | 8 | 35.18 |
| N21 | 6047278 | 805 | 6041 | 112 | 20 | 35.48 |
| N22 | 5686548 | 425 | 5689 | 122 | 11 | 35.1 |
| N23 | 6246356 | 279 | 6250 | 105 | 7 | 34.76 |
| N24 | 6083667 | 332 | 6065 | 100 | 7 | 34.92 |
| N25 | 6552745 | 545 | 6375 | 96 | 6 | 34.68 |
| N26 | 6346459 | 979 | 6410 | 101 | 7 | 35.18 |
| N27 | 6484007 | 279 | 6360 | 91 | 6 | 34.67 |
| N28 | 5996583 | 320 | 6003 | 102 | 10 | 35,13 |
| N29 | 5603679 | 484 | 5593 | 95 | 11 | 35.18 |
| N30 | 5706629 | 631 | 5665 | 99 | 9 | 35.3 |
| N31 | 5996429 | 274 | 5885 | 94 | 11 | 34.98 |
| N33 | 5528708 | 290 | 5550 | 109 | 14 | 35.32 |
| N34 | 5640513 | 173 | 5616 | 92 | 9 | 35.28 |
| NR | Country | Phylogenetic Group (panC) | MLST Profile | glp | gmk | ilv | pta | pur | pyc | tpi |
|---|---|---|---|---|---|---|---|---|---|---|
| N1 | Lithuania | clade 4 | ? | 14 | 8 * | 48 | 45 | 58 | 51 | 7 |
| N2 | Lithuania | clade 4 | ? | 285 | 8 | 283 * | 45 * | 58 | 87 | 7 |
| N3 | Tunesia | clade 5 | 223 | 43 | 26 | 35 | 42 | 39 | 41 | 63 |
| N4 | Tunesia | clade 4 | 1009 | 16 | 6 | 170 | 9 | 4 | 7 | 21 |
| N5 | Nederlands | clade 2 | 616 | 81 | 53 | 117 | 71 | 113 | 93 | 80 |
| N6 | Nederlands | clade 2 | ? * | 110 | 56 | 111* | 188 | 64 | 96 | 26 |
| N7 | Slovenia | clade 6 | 434 | 26 | 21 | 126 | 104 | 78 | 32 | 18 |
| N8 | Slovenia | clade 6 | ? * | 274 | 115 | 227 | 200 | 195 | 170 | 163 * |
| N9 | Switzerland | clade 6 | 428 | 108 | 51 | 130 | 121 | 109 | 92 | 79 |
| N10 | Switzerland | clade 6 | 434 | 26 | 21 | 126 | 104 | 78 | 32 | 18 |
| N11 | Hungary | clade 5 | ? * | 87 | 26 * | 78 | 90 | 273 | 41 | 30 |
| N12 | Hungary | clade 5 | ? | 87 | 26 | 78 | 90 | 273 | 220 | 30 |
| N13 | Hungary | clade 4 | 23 | 15 | 7 | 7 | 2 | 5 | 8 | 13 |
| N14 | Hungary | clade 2 | ? * | 118 * | 35 | 171 * | 22 | 96 | 20 | 26 |
| N15 | Serbia | clade 5 | 72 * | 43 | 26 * | 35 | 40 | 39 | 41 | 30 |
| N16 | Serbia | clade 3 | 1766 * | 229 * | 5 | 271 | 240 | 281 | 183 | 190 |
| N17 | Serbia | clade 5 | 278 | 83 | 26 | 35 | 42 | 39 | 71 | 30 |
| N18 | Serbia | clade 5 | 1484 * | 43 | 26 | 78 | 42 | 39 * | 71 | 30 * |
| N19 | Turkey | clade 6 | ? * | 75 * | 104 * | 303 * | 15 * | 103 * | 202 * | 128 * |
| N20 | Turkey | clade 4 | 56 | 15 | 7 | 7 | 2 | 7 | 26 | 13 |
| N21 | Turkey | clade 1 | ? * | 134 | 13 | 274 | 253 | 84 | 44 | 35 * |
| N22 | Turkey | clade 4 | 4 | 13 | 8 | 8 | 11 | 11 | 12 | 7 |
| N23 | Turkey | clade 4 | 12 | 15 | 7 | 7 | 2 | 7 | 10 | 13 |
| N24 | Turkey | clade 5 | 886 | 43 | 50 | 35 | 125 | 70 | 41 | 30 |
| N25 | Turkey | clade 4 | 12 | 15 | 7 | 7 | 2 | 7 | 10 | 13 |
| N26 | Turkey | clade 4 | 56 | 15 | 7 | 7 | 2 | 7 | 26 | 13 |
| N27 | Turkey | clade 4 | 12 | 15 | 7 | 7 | 2 | 7 | 10 | 13 |
| N28 | Turkey | clade 6 | ? | 8 | 10 | 22 | 15 | 35 | 9 | 11 |
| N29 | Denmark | clade 6 | 635 * | 26 | 21 | 104 * | 68 | 27* | 32 | 18 |
| N30 | Denmark | clade 6 | ? * | 108 | 139 | 104 | 15 * | 221 | 92 | 79 |
| N31 | Austria | clade 5 | 487 | 83 | 26 | 143 | 133 | 91 | 41 | 30 |
| N33 | Belgium | clade 6 | 254 | 103 | 63 | 5 | 15 | 94 | 70 | 11 |
| N34 | Belgium | clade 6 | 734 | 25 | 10 | 22 | 165 | 83 | 23 | 11 |
| Isolate | Country | Phylogeny | Traditional | 16S RNA gene | Species ANI | Species DNA/DNA |
|---|---|---|---|---|---|---|
| N21 | Turkey | clade 1 | B. cereus | B. cereus | B. pseudomycoides | B pseudomycoides |
| N14 | Hungary | clade 2 | B. cereus | B. cereus | B. mosaicus | B toyonensis |
| N16 | Serbia | clade 2 | B. cereus | B. cereus | B. mosaicus | New species 2 |
| N5 | Nederlands | clade 2 | B. cereus | B. cereus | B. mosaicus | New species 3 |
| N6 | Nederlands | clade 2 | B. cereus | B. cereus | B. mosaicus | New species 5 |
| N1 | Lithuania | clade 4 | B. cereus | B. cereus | B. cereus s.s. biovar Thuringiensis; B. thuringiensis | New species 1 |
| N13 | Hungary | clade 4 | B. cereus | B. cereus | B. cereus s.s. | B. wiedmanii |
| N2 | Lithuania | clade 4 | B. cereus | B. cereus | B. cereus s.s. biovar Thuringiensis; B. thuringiensis | New species 1 |
| N20 | Turkey | clade 4 | B. cereus | B. cereus | B. cereus s.s. | New species 7 |
| N22 | Turkey | clade 4 | B. cereus | B. cereus | B. cereus s.s. | B cereus |
| N23 | Turkey | clade 4 | B. cereus | B. mycoides | B. cereus s.s. | New species 7 |
| N25 | Turkey | clade 4 | B. cereus | B. mycoides | B. cereus s.s. biovar Thuringiensis; B. thuringiensis | New species 7 |
| N26 | Turkey | clade 4 | B. cereus | B. mycoides | B. cereus s.s. | New species 7 |
| N27 | Turkey | clade 4 | B. cereus | B. mycoides | B. cereus s.s. biovar Thuringiensis; B. thuringiensis | New species 7 |
| N4 | Tunesia | clade 4 | B. cereus | B. mycoides | B. cereus s.s. | Bacillus cereus |
| N11 | Hungary | clade 5 | B. cereus | B. mycoides | B. toyonensis | B toyonensis |
| N12 | Hungary | clade 5 | B. cereus | B. mycoides | B. toyonensis | B. toyonensis |
| N15 | Serbia | clade 5 | B. cereus | B. mycoides | B. toyonensis | B.toyonensis |
| N17 | Serbia | clade 5 | B. cereus | B. pseudomycoides | B. toyonensis | B toyonensis |
| N18 | Serbia | clade 5 | B. cereus | B. sp | B. toyonensis | B toyonensis |
| N24 | Turkey | clade 5 | B. cereus | B. sp. | B. toyonensis | B toyonensis |
| N3 | Tunesia | clade 5 | B. cereus | B. thuringiensis | B. toyonensis | B. toyonensis |
| N31 | Austria | clade 5 | B. cereus | B. thuringiensis | B. toyonensis | B toyonensis |
| N10 | Switzerland | clade 6 | B. weihenstephanensis | B. toyonensis | B. mycoides | B toyonensis |
| N19 | Turkey | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | New species 6 |
| N28 | Turkey | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | B. mycoides |
| N29 | Denmark | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | New species 4 |
| N30 | Denmark | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | New species 4 |
| N33 | Belgium | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | B mycoides |
| N34 | Belgium | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | B mycoides |
| N7 | Slovenia | clade 6 | B. weihenstephanensis | B. wiedmanii | B. mycoides | B. proteolyticus |
| N8 | Slovenia | clade 6 | B. weihenstephanensis | Not identified | B. mycoides | Bacillus proteolyticus |
| N9 | Switzerland | clade 6 | B. weihenstephanensis | B. mycoides | B. mycoides | New species 4 |
| Isolate | Group | cerA | cerB | clo | cytK2 | entA | entFM | hblA | hblB | hblC | hblcD | hlyII | inhA1 | inhA2 | nheA | nheB | nheC | plcA | plcB | sph |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N21 | I | 89 | 0 | 0 | 0 | 91 | 71 | 89 | 0 | 85 | 88 | 0 | 83 | 86 | 74 | 86 | 79 | 0 | 89 | 0 |
| N14 | II | 95 | 90 | 98 | 0 | 96 | 94 | 95 | 87 | 94 | 99 | 0 | 94 | 96 | 96 | 100 | 95 | 94 | 95 | 98 |
| N16 | II | 95 | 88 | 96 | 0 | 94 | 92 | 99 | 96 | 99 | 100 | 96 | 94 | 96 | 97 | 100 | 94 | 96 | 95 | 96 |
| N5 | II | 96 | 89 | 99 | 0 | 94 | 95 | 95 | 86 | 94 | 100 | 96 | 93 | 97 | 97 | 100 | 95 | 94 | 96 | 97 |
| N6 | II | 95 | 90 | 95 | 0 | 97 | 95 | 98 | 87 | 95 | 99 | 95 | 94 | 96 | 97 | 99 | 95 | 94 | 95 | 98 |
| N1 | IV | 100 | 94 | 99 | 98 | 98 | 97 | 99 | 98 | 98 | 100 | 0 | 98 | 99 | 99 | 100 | 99 | 95 | 100 | 91 |
| N13 | IV | 100 | 95 | 99 | 0 | 95 | 95 | 99 | 98 | 98 | 100 | 99 | 94 | 99 | 100 | 100 | 98 | 96 | 100 | 91 |
| N2 | IV | 100 | 94 | 99 | 99 | 97 | 92 | 99 | 98 | 98 | 100 | 0 | 98 | 99 | 99 | 100 | 98 | 95 | 100 | 90 |
| N20 | IV | 100 | 98 | 99 | 0 | 95 | 95 | 99 | 98 | 98 | 100 | 99 | 94 | 99 | 100 | 100 | 98 | 96 | 100 | 90 |
| N22 | IV | 100 | 95 | 99 | 100 | 99 | 97 | 99 | 99 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 100 | 100 | 92 |
| N23 | IV | 100 | 95 | 99 | 0 | 95 | 93 | 99 | 98 | 98 | 100 | 99 | 94 | 99 | 100 | 100 | 98 | 96 | 100 | 91 |
| N25 | IV | 100 | 95 | 99 | 97 | 95 | 94 | 99 | 98 | 98 | 100 | 99 | 94 | 98 | 100 | 100 | 98 | 96 | 100 | 91 |
| N26 | IV | 100 | 0 | 99 | 0 | 95 | 95 | 99 | 98 | 98 | 100 | 99 | 94 | 99 | 100 | 100 | 98 | 96 | 100 | 90 |
| N27 | IV | 100 | 95 | 99 | 97 | 95 | 94 | 99 | 98 | 98 | 100 | 99 | 94 | 98 | 100 | 100 | 98 | 96 | 100 | 91 |
| N4 | IV | 100 | 95 | 99 | 99 | 98 | 95 | 99 | 98 | 99 | 100 | 99 | 94 | 99 | 99 | 100 | 98 | 97 | 100 | 92 |
| N11 | V | 98 | 90 | 91 | 0 | 97 | 90 | 93 | 86 | 94 | 99 | 0 | 96 | 97 | 97 | 100 | 93 | 90 | 98 | 94 |
| N12 | V | 99 | 89 | 92 | 0 | 95 | 90 | 92 | 86 | 94 | 99 | 0 | 96 | 97 | 97 | 100 | 94 | 90 | 99 | 93 |
| N15 | V | 98 | 89 | 93 | 0 | 96 | 90 | 95 | 85 | 95 | 99 | 0 | 94 | 97 | 96 | 100 | 94 | 90 | 98 | 93 |
| N17 | V | 99 | 89 | 0 | 0 | 97 | 90 | 95 | 86 | 95 | 100 | 0 | 94 | 97 | 96 | 100 | 94 | 90 | 99 | 93 |
| N18 | V | 99 | 89 | 0 | 0 | 97 | 90 | 95 | 86 | 95 | 100 | 0 | 94 | 97 | 96 | 100 | 94 | 90 | 99 | 93 |
| N24 | V | 99 | 89 | 93 | 0 | 97 | 90 | 95 | 86 | 95 | 99 | 0 | 94 | 97 | 96 | 100 | 94 | 91 | 99 | 93 |
| N3 | V | 98 | 89 | 93 | 0 | 96 | 90 | 95 | 86 | 95 | 99 | 0 | 94 | 97 | 96 | 100 | 94 | 90 | 98 | 93 |
| N31 | V | 99 | 89 | 93 | 98 | 97 | 90 | 95 | 86 | 94 | 99 | 0 | 95 | 97 | 97 | 100 | 93 | 91 | 99 | 93 |
| N10 | VI | 98 | 89 | 97 | 0 | 88 | 88 | 92 | 87 | 84 | 94 | 0 | 91 | 93 | 94 | 99 | 93 | 93 | 98 | 92 |
| N19 | VI | 98 | 87 | 89 | 0 | 88 | 90 | 69 | 65 | 79 | 0 | 0 | 91 | 94 | 96 | 100 | 94 | 95 | 98 | 92 |
| N28 | VI | 95 | 87 | 97 | 0 | 88 | 87 | 97 | 88 | 93 | 98 | 0 | 91 | 95 | 97 | 100 | 92 | 0 | 95 | 90 |
| N29 | VI | 97 | 88 | 0 | 0 | 88 | 89 | 91 | 87 | 84 | 94 | 0 | 93 | 96 | 95 | 100 | 93 | 92 | 97 | 91 |
| N30 | VI | 98 | 89 | 98 | 0 | 88 | 88 | 90 | 87 | 88 | 96 | 0 | 91 | 94 | 96 | 100 | 93 | 93 | 98 | 92 |
| N33 | VI | 95 | 87 | 98 | 0 | 94 | 88 | 91 | 89 | 89 | 93 | 0 | 91 | 93 | 97 | 100 | 93 | 93 | 95 | 91 |
| N34 | VI | 95 | 87 | 95 | 0 | 93 | 88 | 98 | 87 | 93 | 99 | 0 | 91 | 95 | 95 | 98 | 94 | 93 | 95 | 91 |
| N7 | VI | 98 | 89 | 97 | 0 | 88 | 88 | 92 | 87 | 84 | 94 | 0 | 91 | 93 | 94 | 99 | 93 | 93 | 98 | 92 |
| N8 | VI | 91 | 88 | 98 | 0 | 88 | 85 | 91 | 87 | 84 | 92 | 0 | 91 | 92 | 95 | 99 | 94 | 0 | 91 | 89 |
| N9 | VI | 98 | 87 | 98 | 0 | 87 | 89 | 90 | 89 | 88 | 96 | 0 | 91 | 93 | 96 | 99 | 93 | 92 | 98 | 92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zervas, A.; Aggerbeck, M.R.; Allaga, H.; Güzel, M.; Hendriks, M.; Jonuškienė, I.; Kedves, O.; Kupeli, A.; Lamovšek, J.; Mülner, P.; et al. Identification and Characterization of 33 Bacillus cereus sensu lato Isolates from Agricultural Fields from Eleven Widely Distributed Countries by Whole Genome Sequencing. Microorganisms 2020, 8, 2028. https://doi.org/10.3390/microorganisms8122028
Zervas A, Aggerbeck MR, Allaga H, Güzel M, Hendriks M, Jonuškienė I, Kedves O, Kupeli A, Lamovšek J, Mülner P, et al. Identification and Characterization of 33 Bacillus cereus sensu lato Isolates from Agricultural Fields from Eleven Widely Distributed Countries by Whole Genome Sequencing. Microorganisms. 2020; 8(12):2028. https://doi.org/10.3390/microorganisms8122028
Chicago/Turabian StyleZervas, Athanasios, Marie Rønne Aggerbeck, Henrietta Allaga, Mustafa Güzel, Marc Hendriks, IIona Jonuškienė, Orsolya Kedves, Ayse Kupeli, Janja Lamovšek, Pascal Mülner, and et al. 2020. "Identification and Characterization of 33 Bacillus cereus sensu lato Isolates from Agricultural Fields from Eleven Widely Distributed Countries by Whole Genome Sequencing" Microorganisms 8, no. 12: 2028. https://doi.org/10.3390/microorganisms8122028