Assessing the Multivariate Relationship between the Human Infant Intestinal Exfoliated Cell Transcriptome (Exfoliome) and Microbiome in Response to Diet

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Isolation and Analysis of Stool Microbial DNA and Host PolyA+ mRNA
2.3. Data Normalization, Transformation and Prior Knowledge Lists
2.4. Differential Gene Expression Analysis
2.5. Quantification of Fecal Volatile Fatty Acids
2.6. Gut Metagenome and Host Transcriptome Multivariate Analyses and Data Integration
2.7. Data Deposition
3. Results
3.1. Fecal Volatile Fatty Acid Concentrations
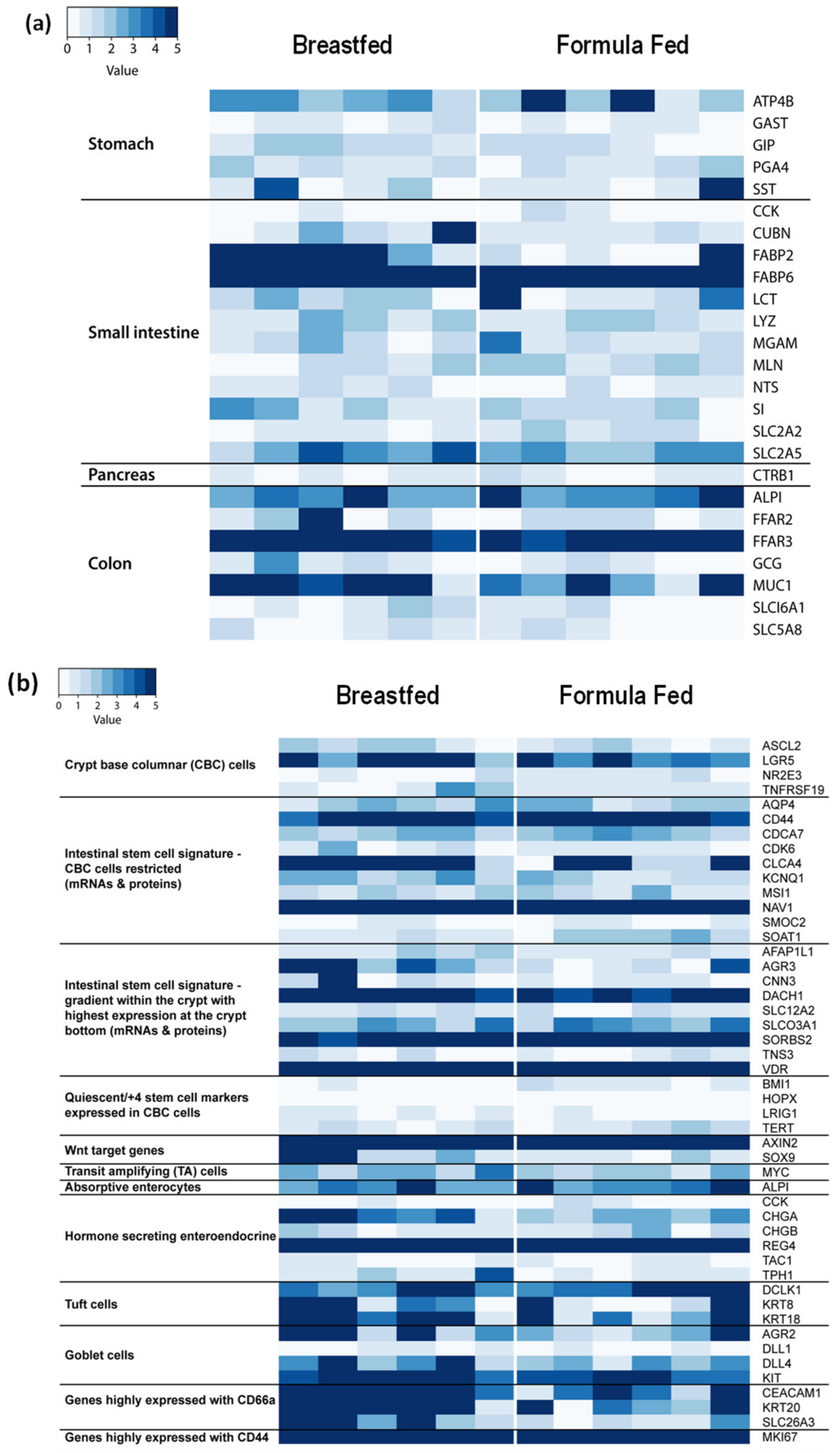
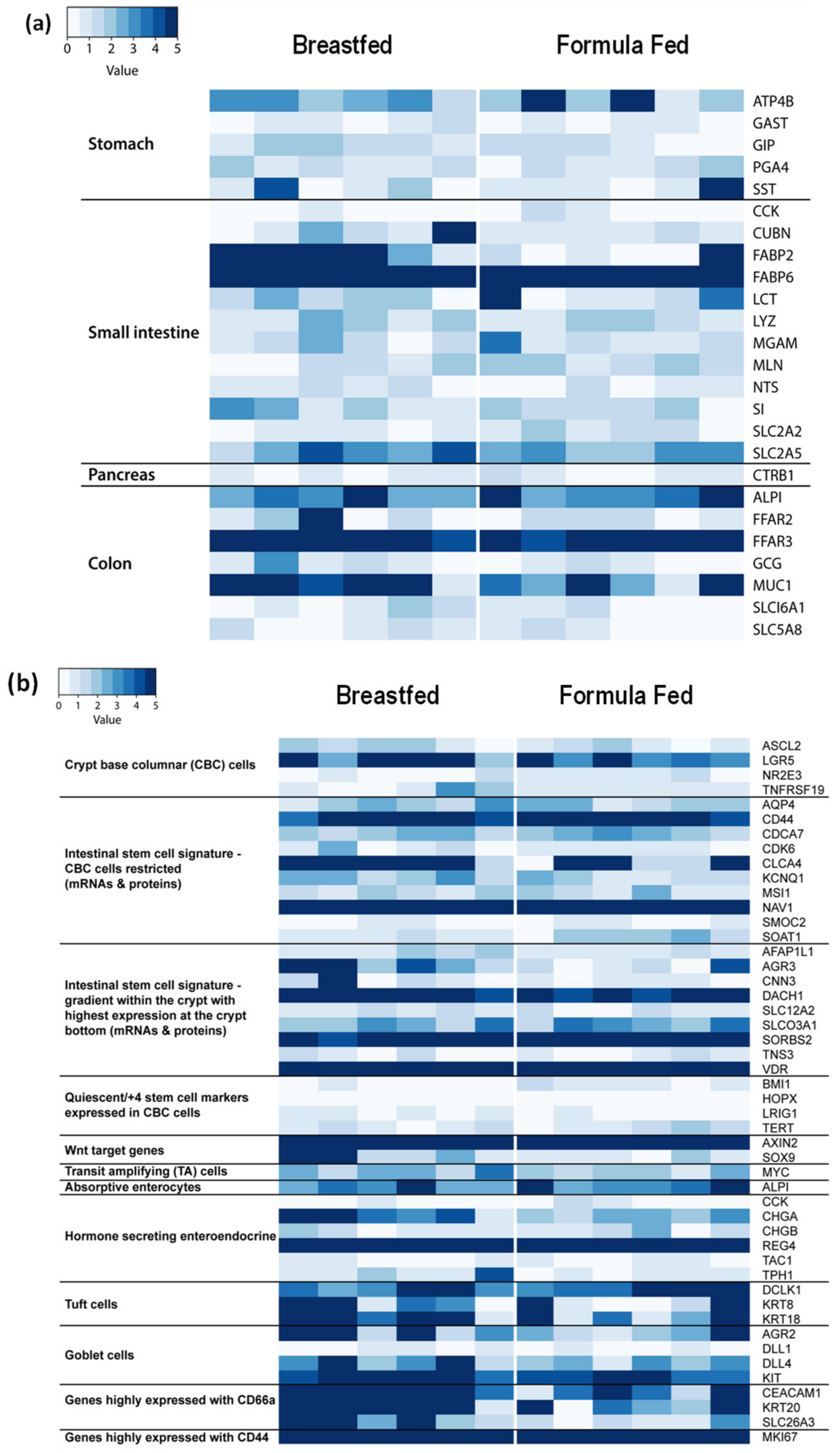
3.2. Anatomic Origin of Exfoliated Intestinal Epithelial Cells
3.3. Data Structure and Interactions between the Host Transcriptome and Gut Microbiome in Breast- and Formula-Fed Neonates
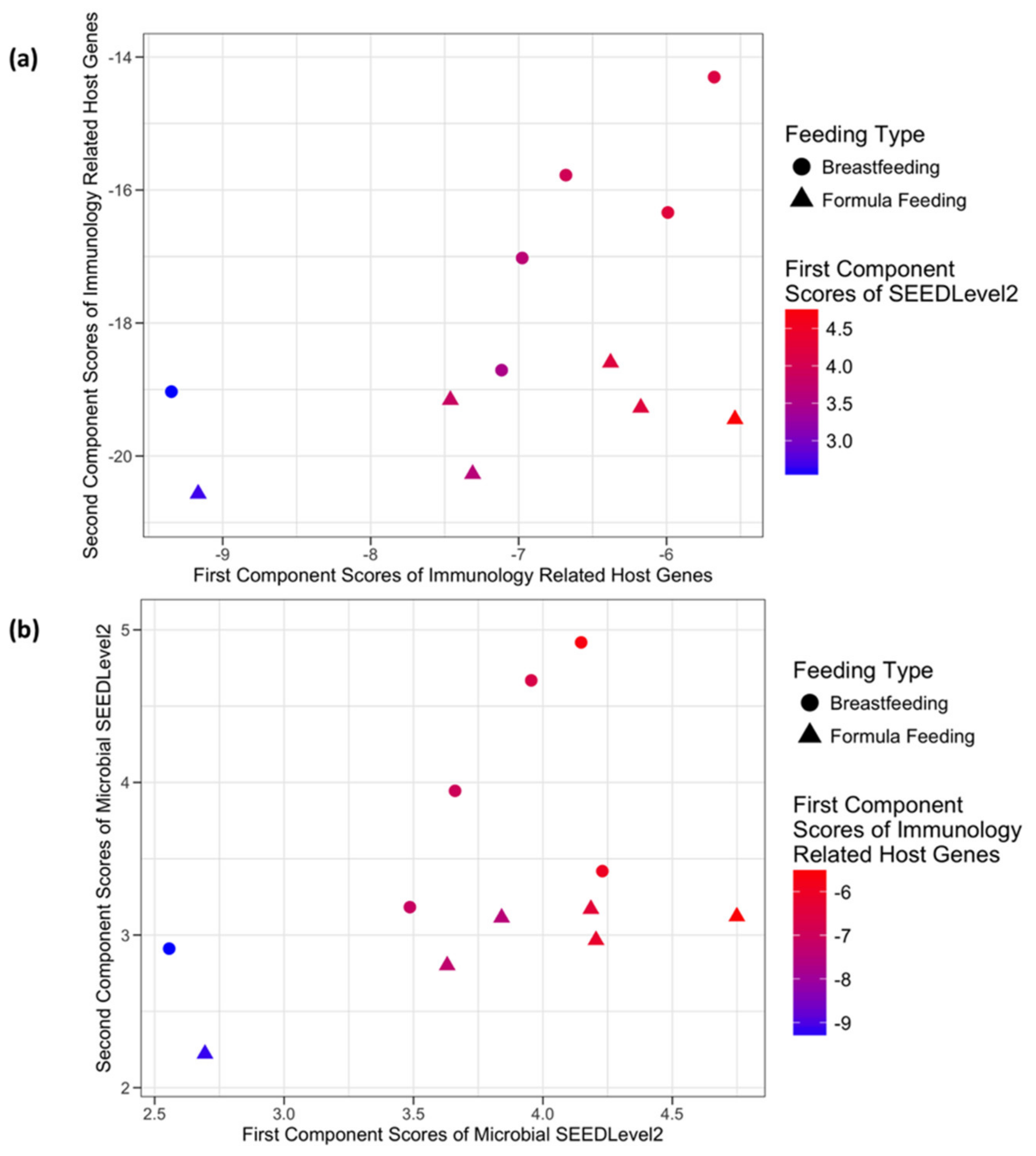
3.3.1. SCFA Signaling Genes and Microbial SEEDLevel2 Categories
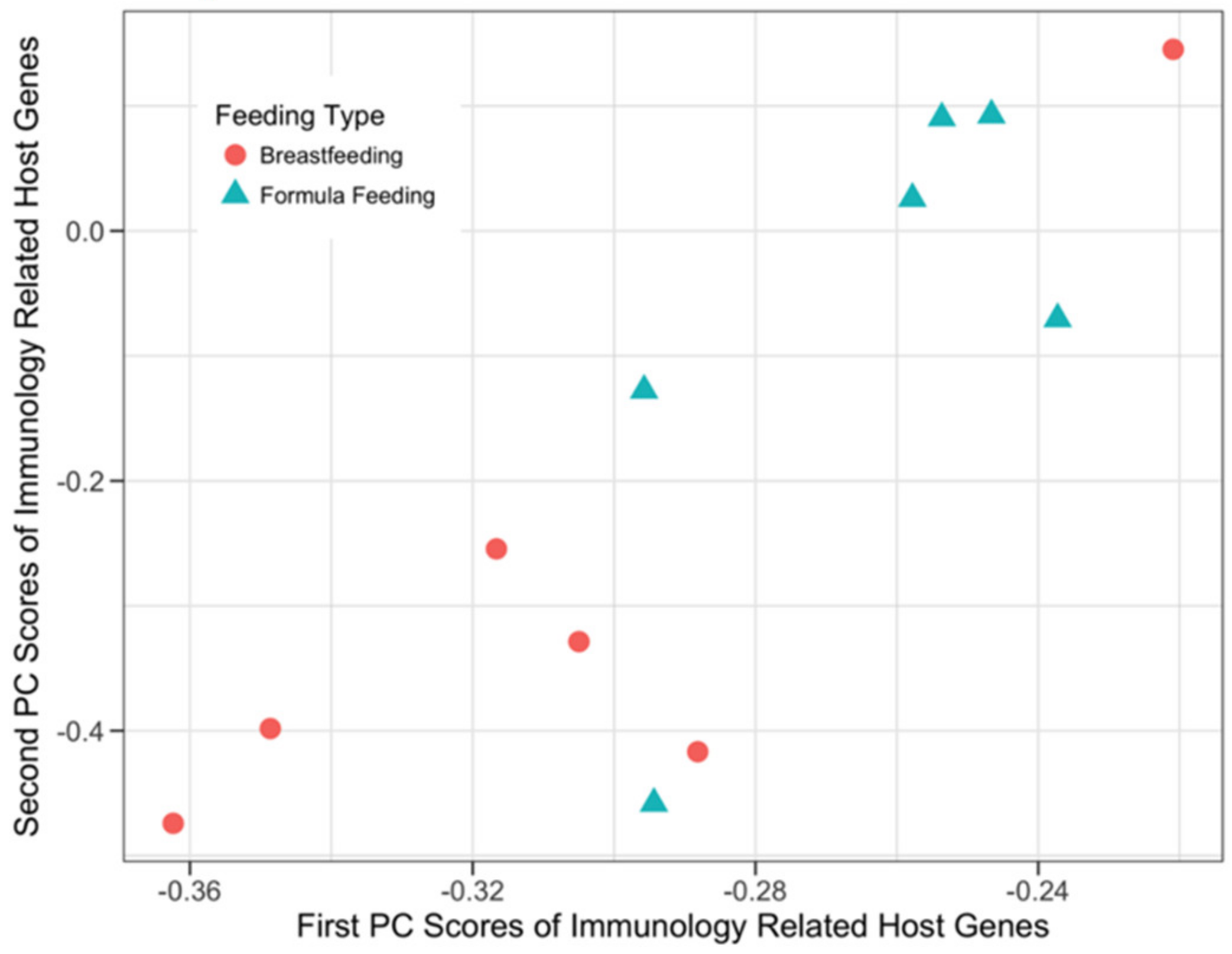
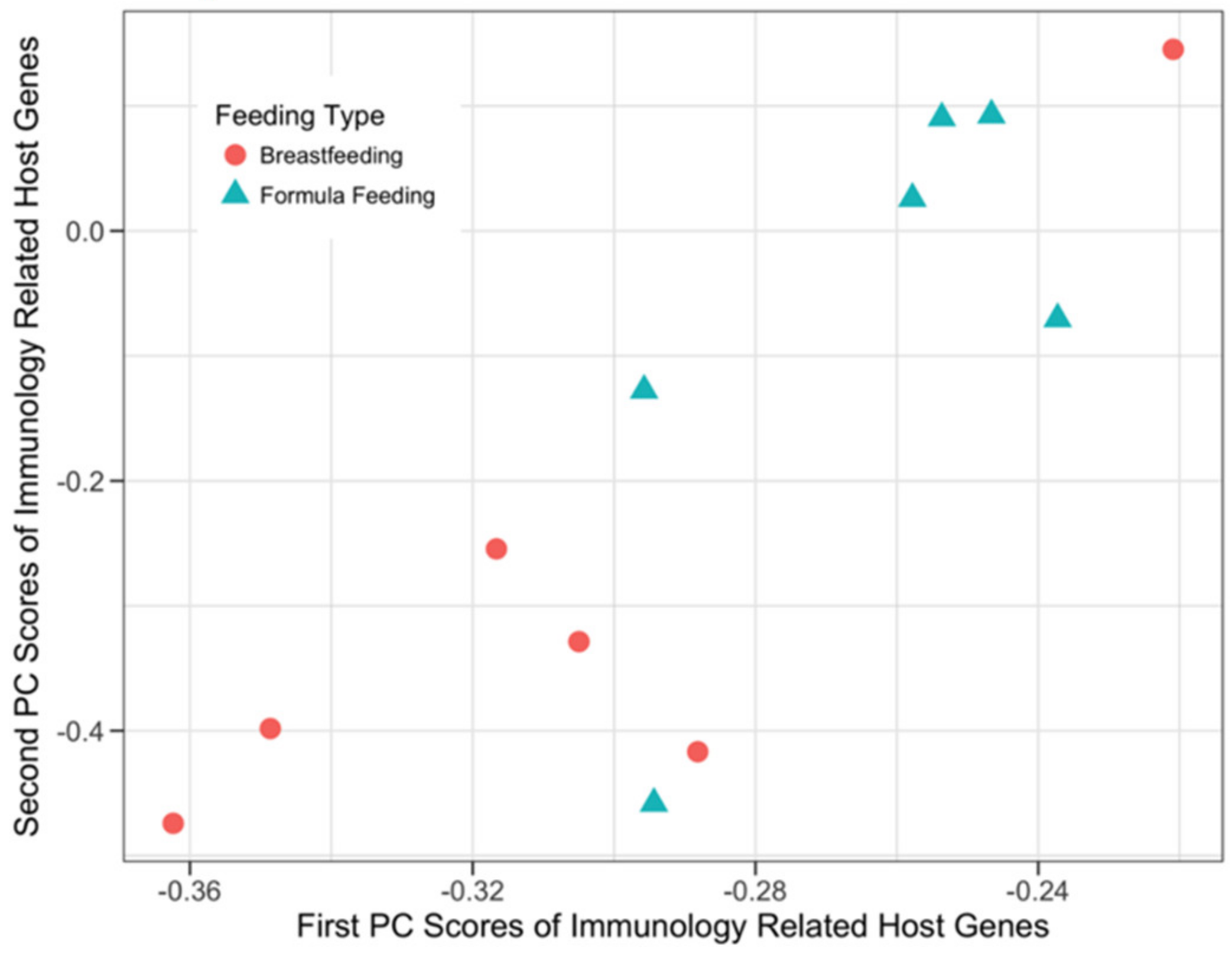
3.3.2. Host Immunology and Defense Genes and Microbial SEEDLevel2 Categories
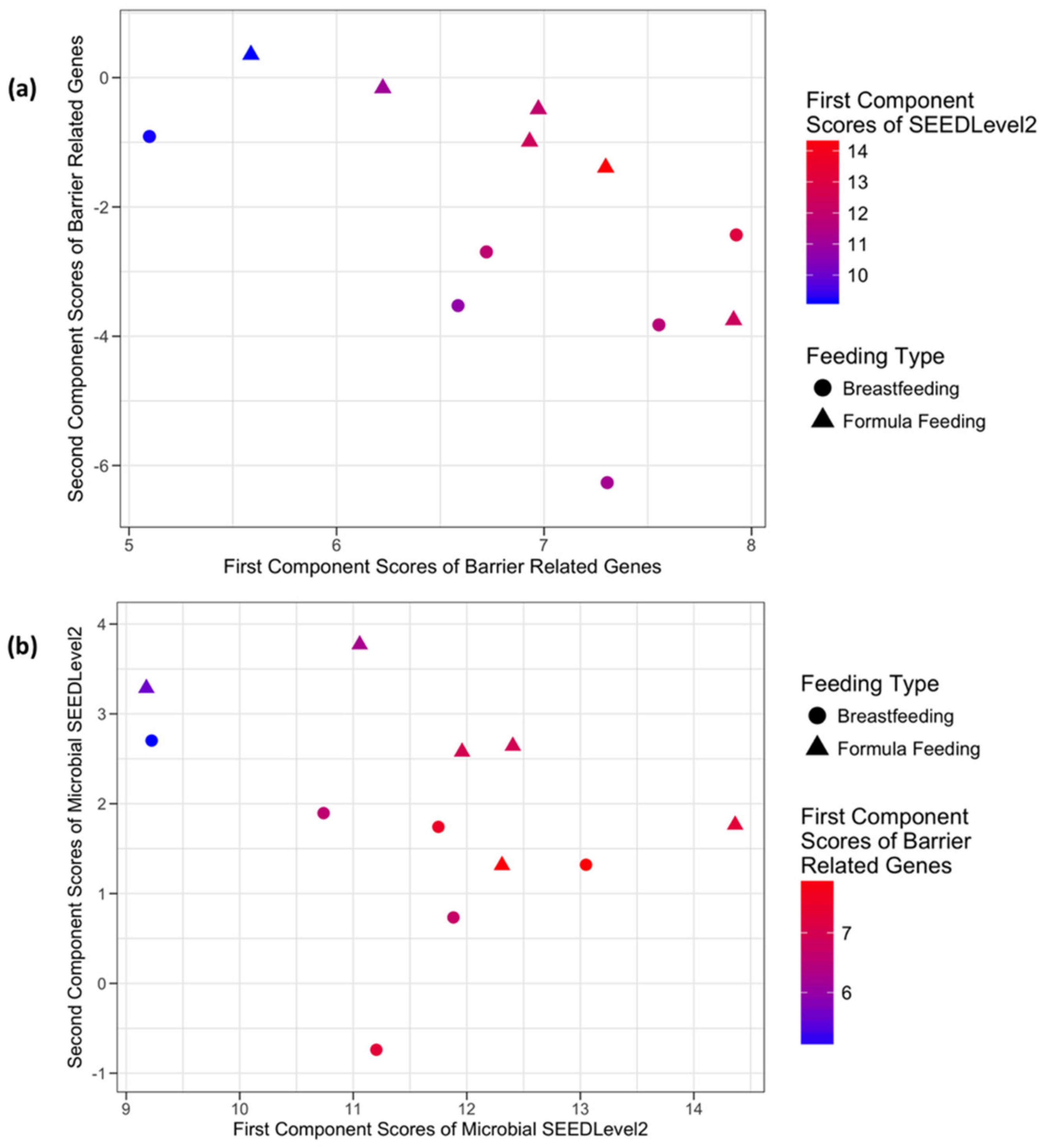
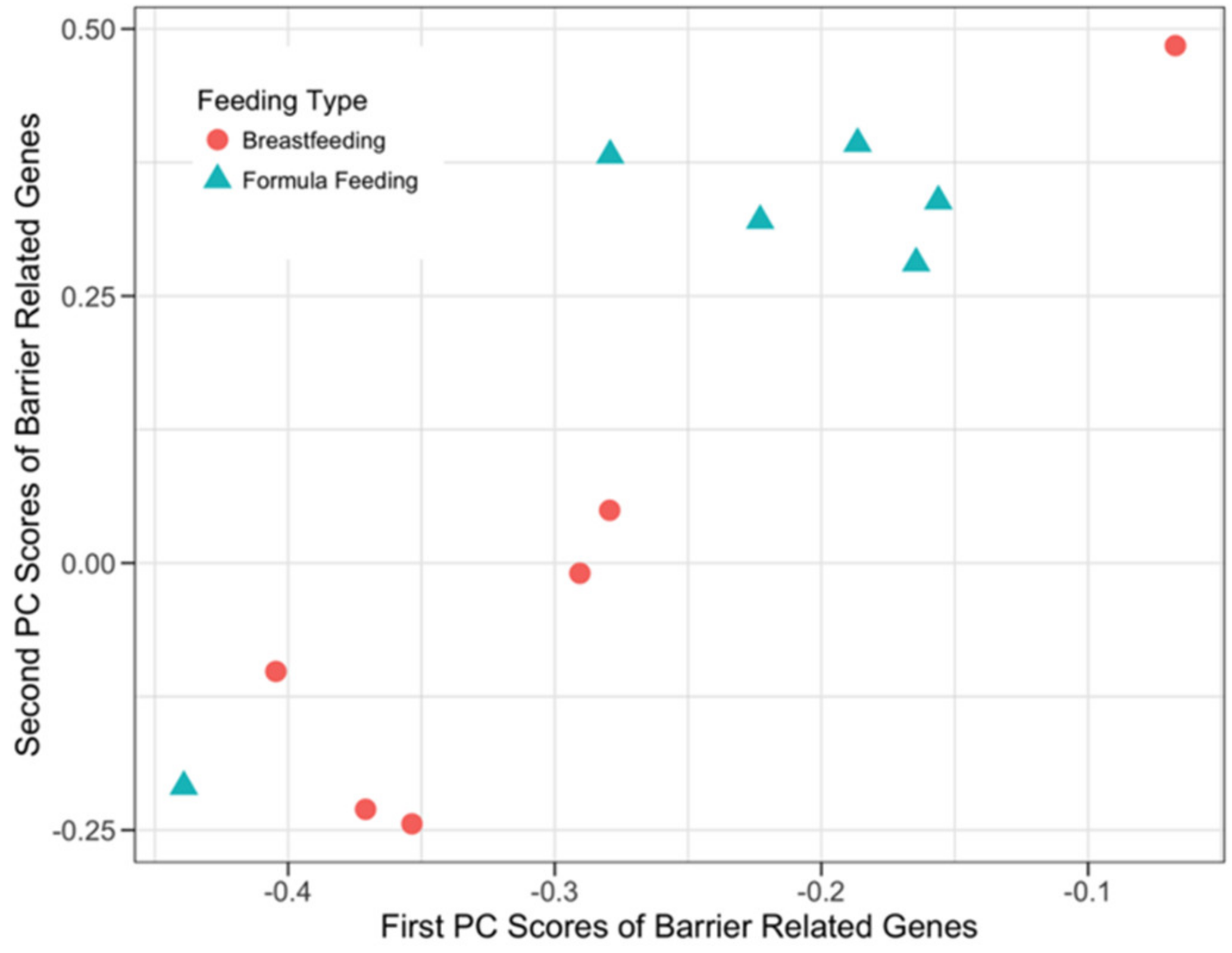
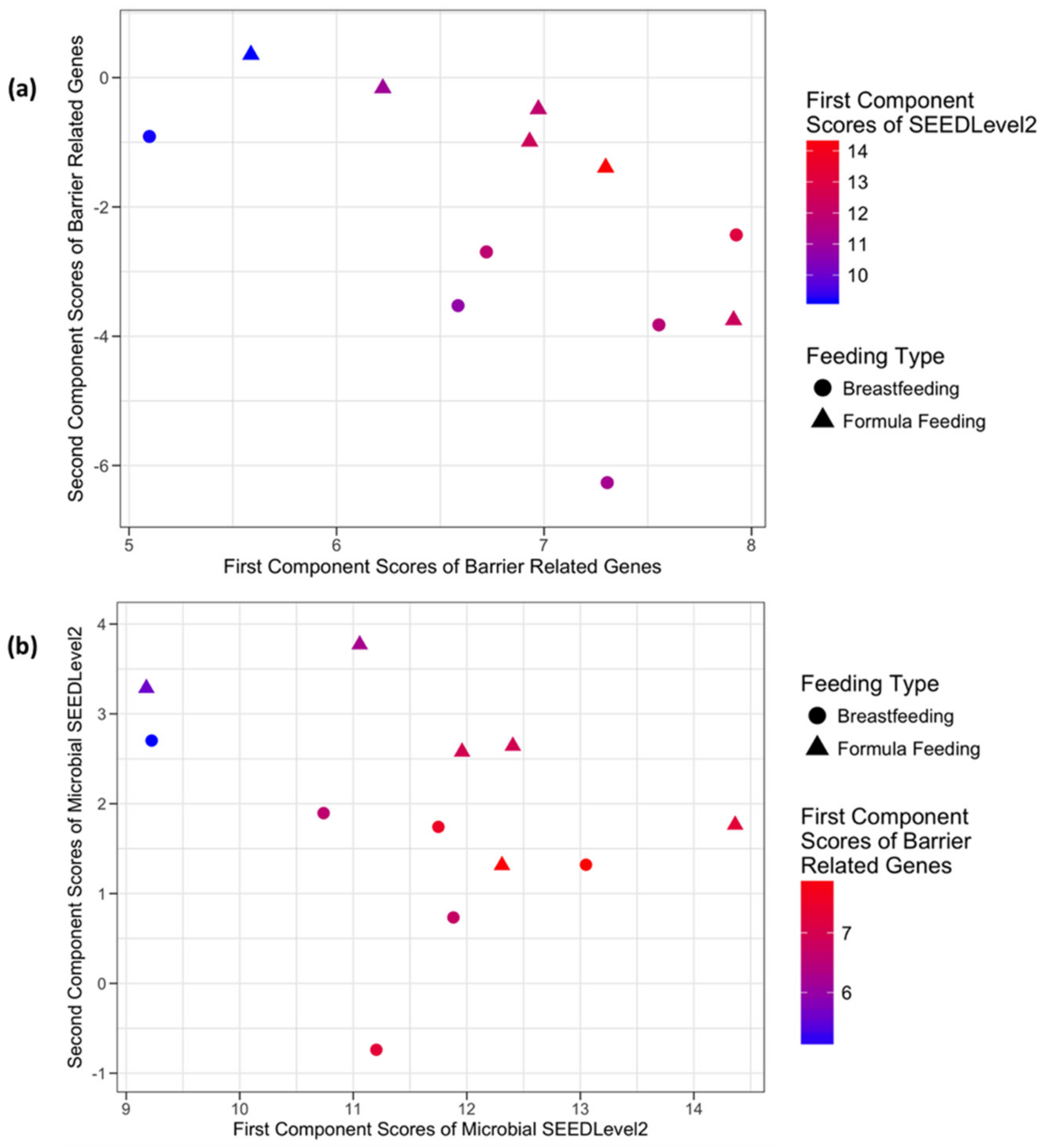
3.3.3. Host Barrier Function-Related Genes and Microbial SEEDLevel2 Categories
3.4. Differential Gene Expression
4. Discussion
4.1. Anatomical Source of Exfoliated Cells
4.2. Data Structure Detected by sPCA
4.3. Correlative Data Structure Detected by sCCA
4.4. Description of Genes Identified by Multivariate Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix A.1. The General Settings of PCA, CCA, sPCA and sCCA
Appendix A.2. Synthetically Generated Data Analysis to Compare the Performance of sCCA, sPCA, and Sub-Dimensional CCA
References
- Rakoff-Nahoum, S.; Kong, Y.; Kleinstein, S.H.; Subramanian, S.; Ahern, P.P. Analysis of gene–environment interactions in postnatal development of the mammalian intestine. Proc. Natl. Acad. Sci. USA 2015, 112, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Walker, W.A. Diet and host-microbial crosstalk in postnatal intestinal immune homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 14–25. [Google Scholar] [CrossRef]
- Chapkin, R.S.; Zhao, C.; Ivanov, I.; Davidson, L.A.; Goldsby, J.S.; Lupton, J.R.; Mathai, R.A.; Monaco, M.H.; Rai, D.; Russell, W.M.; et al. Noninvasive stool-based detection of infant gastrointestinal development using gene expression profiles from exfoliated epithelial cells. Am. J. Physiol. Gastrointest. Liver. Physiol. 2010, 298, G582–G589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.M.; Davidson, L.A.; Herman, D.; Martin, C.R.; Goldsby, J.S.; Ivanov, I.V.; Donovan, S.M.; Chapkin, R.S. Non-invasive analysis of intestinal development in preterm and term infants using RNA-sequencing. Sci. Rep. 2014, 4, 5453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potten, C.S.; Schofield, R.; Lajtha, L.G. A comparison of cell replacement in bone marrow, testis and three regions of epithelium. Biochim. Biophys. Acta. 1979, 560, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Kaeffer, B. Survival of exfoliated epithelial cells: A delicate balance between anoikis and apoptosis. J. Biomed. Biotechnol. 2011, 534139. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar]
- Kaeffer, B.; des Robert, C.; Alexandre-Gouabau, M.C.; Pagniez, A.; Legrand, A.; Amarger, V.; Küster, A.; Piloquet, H.; Champ, M.; le Huërou-Luron, I.; et al. Recovery of exfoliated cells from the gastrointestinal tract of premature infants: A new tool to perform “noninvasive biopsies?”. Pediatr. Res. 2007, 62, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Holland, N.; Harmatz, P.; Golden, D.; Hubbard, A.; Wu, Y.Y.; Bae, J.; Chen, C.; Huen, K.; Heyman, M.B. Cytogenetic damages in blood lymphocytes and exfoliated epithelial cells of children with inflammatory bowel disease. Pediatr. Res. 2007, 61, 209–214. [Google Scholar] [CrossRef]
- Kaeffer, B.; Legrand, A.; Moyon, T.; Frondas-Chauty, A.; Billard, H.; Guzman-Quevedo, O.; Darmaun, D.; Rozé, J.C. Non-invasive exploration of neonatal gastric epithelium by using exfoliated epithelial cells. PLoS ONE 2011, 6, e25562. [Google Scholar] [CrossRef]
- Whitfield-Cargile, C.M.; Cohen, N.D.; He, K.; Ivanov, I.; Goldsby, J.S.; Chamoun-Emanuelli, A.; Weeks, B.R.; Davidson, L.A.; Chapkin, R.S. The non-invasive exfoliated transcriptome (exfoliome) reflects the tissue-level transcriptome in a mouse model of NSAID enteropathy. Sci. Rep. 2017, 7, 14687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, S.M.; Wang, M.; Monaco, M.H.; Martin, C.R.; Davidson, L.A.; Ivanov, I.; Chapkin, R.S. Noninvasive molecular fingerprinting of host-microbe interactions in neonates. FEBS Letts. 2014, 588, 4112–4119. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, M.; Wu, S.; Lebrilla, C.B.; Chapkin, R.S.; Ivanov, I.; Donovan, S.M. Fecal microbiota composition of breast-fed infants is correlated with human milk oligosaccharides consumed. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 825–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Friedberg, I.; Ivanov, I.V.; Davidson, L.A.; Goldsby, J.S.; Dahl, D.B.; Herman, D.; Wang, M.; Donovan, S.M.; Chapkin, R.S. A metagenomic study of diet-dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biol. 2012, 13, R32. [Google Scholar] [CrossRef] [Green Version]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [Green Version]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Witten, D.M.; Tibshirani, R.J. Extensions of sparse canonical correlation analysis with applications to genomic data. Stat. Appl. Genet. Mol. Biol. 2009, 8, 28. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Bittner, M.L.; Chapkin, R.S.; Dougherty, E.R. Characterization of the effectiveness of reporting lists of small feature sets relative to the accuracy of the prior biological knowledge. Cancer Inform. 2010, 9, 49–60. [Google Scholar]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, J.N. metagenomeSeq: Statistical Analysis for Sparse High-Throughput Sequencing. Bioconductor 2020. Available online: https://rdrr.io/bioc/metagenomeSeq/man/metagenomeSeq-package.html (accessed on 1 June 2019).
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Amaratunga, D.; Cabrera, J. Analysis of data from viral DNA microchips. J. Am. Stat. Assoc. 2001, 96, 1161–1170. [Google Scholar] [CrossRef]
- Ritchie, M.E. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Li, M.; Bauer, L.L.; Chen, X.; Wang, M.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Fahey, G.C., Jr.; Donovan, S.M. Microbial composition and in vitro fermentation patterns of human milk oligosaccharides differ between formula-fed and sow-reared piglets. J. Nutr. 2012, 142, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Krzanowski, W. Principles of Multivariate Analysis: A User’s Perspective; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Jolliffe, I.T.; Trendafilov, N.T.; Uddin, M. A modified principal component technique based on the LASSO. J. Comp. Graph. Stat. 2003, 12, 531–547. [Google Scholar] [CrossRef] [Green Version]
- Bach, F.R.; Jordan, M.I. A Probabilistic Interpretation of Canonical Correlation Analysis; Technical Report 688; Department of Statistics, University of California: Berkeley, CA, USA, 2005; Available online: https://statistics.berkeley.edu/sites/default/files/tech-reports/688.pdf (accessed on 1 June 2019).
- Lampe, J.W.; Kim, E.; Levy, L.; Davidson, L.A.; Goldsby, J.S.; Miles, F.L.; Navarro, S.L.; Randolph, T.W.; Zhao, N.; Ivanov, I.; et al. Colonic mucosal and exfoliome transcriptomic profiling and fecal microbiome response to a flaxseed lignan extract intervention in humans. Am. J. Clin. Nutr. 2019, 110, 377–390. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Nøhr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Poulsen, S.S.; Han, S.; et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013, 154, 3552–3564. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, J.H.; Newman, T.N.; Oppong, G.O.; Rapsinski, G.J.; Yen, J.H.; Biesecker, S.G.; Wilson, R.P.; Butler, B.P.; Winter, M.G.; Tsolis, R.M.; et al. Microbial amyloids induce interleukin 17A (IL-17A) and IL-22 responses via Toll-like receptor 2 activation in the intestinal mucosa. Infect. Immun. 2012, 80, 4398–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyerich, K.; Dimartino, V.; Cavani, A. IL-17 and IL-22 in immunity: Driving protection and pathology. Eur. J. Immunol. 2017, 47, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef]
- Röhrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human β-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol. 2010, 184, 6688–6694. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Pietsch, D.; Panterodt, T.; Brand, K. Regulation of C/EBPß and the resulting functions in cells of the monocytic lineage. Cell Signal. 2012, 24, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Ngoenkam, J.; Schamel, W.W.; Pongcharoen, S. Selected signalling proteins recruited to the T-cell receptor-CD3 complex. Immunology 2018, 153, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Katrekar, A.; Honigberg, L.A.; Smith, A.M.; Conn, M.T.; Tang, J.; Jeffery, D.; Mortara, K.; Sampang, J.; Williams, S.R.; et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J. Biol. Chem. 2006, 261, 110002–110010. [Google Scholar] [CrossRef] [Green Version]
- Senaldi, G.; Stolina, M.; Guo, J.; Faggioni, R.; McCabe, S.; Kaufman, S.A.; Van, G.; Xu, W.; Fletcher, F.A.; Boone, T.; et al. Regulatory effects of novel neurotrophin-1/b cell-stimulating factor-3 (cardiotrophin-like cytokine) on B cell function. J. Immunol. 2002, 168, 5690–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, S.J.; Linder, A.T.; Brandl, C.; Nitschke, L. B cell siglecs-news on signaling and its interplay with ligand binding. Front. Immunol. 2018, 9, 2820. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Börnchen, C.; Rose, K.; Daubmann, A.; Volksdorf, T.; Wladykowski, E.; Vidal-Y-Sy, S.; Peters, E.M.; Danso, M.; Bouwstra, J.A.; et al. Diverse regulation of claudin-1 and claudin-4 in atopic dermatitis. Am. J. Pathol. 2015, 185, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Skruber, K.; Read, T.A.; Vitriol, E.A. Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell. Sci. 2018, 131, jcs203760. [Google Scholar] [CrossRef] [Green Version]
- Lou, N.; Lennard Richard, M.L.; Yuc, J.; Kindy, M.; Zhang, X.K. The Fli-1 transcription factor is a critical regulator for controlling the expression of chemokine C-X-C motif ligand 2 (CXCL2). Mol. Immunol. 2017, 81, 59–66. [Google Scholar] [CrossRef]
- Parkos, C.A. Neutrophil-epithelial interactions. A double-edged sword. Am. J. Pathol. 2016, 186, 1404–1416. [Google Scholar] [CrossRef] [Green Version]
- Gimeno-Alcañiz, J.V.; Collado, M.C. Impact of human milk on the transcriptomic response of fetal intestinal epithelial cells reveals expression changes of immune-related genes. Food. Funct. 2019, 10, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Yeh, K.Y.; Yeh, M.; Holt, P.R. Induction of intestinal differentiation by systemic and not by luminal corticosterone in adrenalectomized rat pups. Endocrinology 1989, 124, 1898–1904. [Google Scholar] [CrossRef]
- Mahmood, A.; Torres-Pinedo, R. Effect of hormone administration on the sialylation and fucosylation of intestinal microvillus membranes of suckling rats. Pediatr. Res. 1985, 19, 899–902. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Rao, S.D.; Phillips, T.M.; Umbach, D.M.; Bernbaum, J.C.; Archer, J.I.; Rogan, W.J. Are breast-fed infants more resilient? Feeding method and cortisol in infants. J. Pediatr. 2009, 154, 452–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin Neelon, S.E.; Stroo, M.; Mayhew, M.; Maselko, J.; Hoyo, C. Correlation between maternal and infant cortisol varies by breastfeeding status. Infant Behav. Dev. 2015, 40, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanthakumar, N.N.; Young, C.; Ko, J.S.; Meng, D.; Chen, J.; Buie, T.; Walker, W.A. Glucocorticoid responsiveness in developing human intestine: Possible role in prevention of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G85–G92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollanders, J.J.; Heijboer, A.C.; van der Voorn, B.; Rotteveel, J.; Finken, M.J.J. Nutritional programming by glucocorticoids in breast milk: Targets, mechanisms and possible implications. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Grey, K.R.; Davis, E.P.; Sandman, C.A.; Glynn, L.M. Human milk cortisol is associated with infant temperament. Psychoneuroendocrinology 2013, 38, 1178–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dettmer, A.M.; Murphy, A.M.; Guitarra, D.; Slonecker, E.; Suomi, S.J.; Rosenberg, K.L.; Novak, M.A.; Meyer, J.S.; Hinde, K. Cortisol in neonatal mother’s milk predicts later infant social and cognitive functioning in Rhesus monkeys. Child Dev. 2018, 89, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Rifkin, D.B. Latent transforming growth factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta availability. J. Biol. Chem. 2005, 280, 7409–7912. [Google Scholar]
- Urban, Z.; Hucthagowder, V.; Schürmann, N.; Todorovic, V.; Zilberberg, L.; Choi, J.; Sens, C.; Brown, C.W.; Clark, R.D.; Holland, K.E.; et al. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am. J. Hum. Genet. 2009, 85, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Oddy, W.H.; McMahon, R.J. Milk-derived or recombinant transforming growth factor-beta has effects on immunological outcomes: A review of evidence from animal experimental studies. Clin. Exp. Allergy 2011, 41, 783–793. [Google Scholar] [CrossRef]
- Oddy, W.H.; Rosales, F. A systematic review of the importance of milk TGF-beta on immunological outcomes in the infant and young child. Pediatr. Allergy Immunol. 2010, 11, 47–59. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Gallausiaux, C.; Béguet-Crespel, F.; Marinelli, L.; Jamet, A.; Ledue, F.; Blottière, H.M.; Lapaque, N. Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci. Rep. 2018, 8, 9742. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-K.; He, P.; Bialkowska, A.B.; Yang, V.W. SP and KLF transcription factors in digestive physiology and diseases. Gastroenterology 2017, 152, 1845–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley-Freed, W.G.; Efimov, A.; Burnett, P.E.; Short, S.P.; Davis, M.A.; Gumucio, D.L.; Washington, M.K.; Coffey, R.J.; Reynolds, A.B. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J. Clin. Investig. 2010, 120, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.B.; Herbert, L.; Cleveland, J.L.; Berg, S.T.; Gaut, J.R. p120, a novel substrate of protein tyrosine kinase receptors and of p60v-src, is related to cadherin-binding factors beta-catenin, plakoglobin and armadillo. Oncogene 1992, 7, 2439–2445. [Google Scholar]
- Catassi, C.; Bonucci, A.; Coppa, G.V.; Carlucci, A.; Giorgi, P.L. Intestinal permeability changes during the first month: Effect of natural versus artificial feeding. J. Pediatr. Gastroenterol. Nutr. 1995, 21, 383–386. [Google Scholar] [CrossRef]
- Walker, W.A. The dynamic effects of breastfeeding on intestinal development and host defense. Adv. Exp Med. Biol. 2004, 554, 155–170. [Google Scholar]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Chen, J.; Bushman, F.D.; Lewis, J.D.; Wu, G.D.; Li, H. Structure-constrained sparse canonical correlation analysis with an application to microbiome data analysis. Biostatistics 2013, 14, 244–258. [Google Scholar] [CrossRef]
- Ohland, C.L.; Jobin, C. Microbial activities and intestinal homeostasis: A delicate balance between health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Ahern, P.P.; Ridaura, V.K.; Cheng, J.; Gordon, J.I. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci. Transl. Med. 2014, 6, 220ra11. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (µmoles/g Dry Matter) | Breast-Fed (n = 6) | Formula-Fed (n = 6) |
|---|---|---|
| Short Chain Fatty Acids | ||
| Total | 233.2 ± 47.2 | 410.9 ± 37.5 * |
| Acetate | 206.7 ± 51.0 | 327.3 ± 29.7 * |
| Butyrate | 3.17 ± 2.33 | 18.7 ± 8.06 * |
| Propionate | 13.26 ± 4.35 | 64.9 ± 10.9 * |
| Branched Chain Fatty Acids | ||
| Total | 13.35 ± 3.72 | 9.72± 2.42 |
| Isobutyrate | 12.70 ± 3.86 | 3.89 ± 0.98 * |
| Isovalerate | 0.65 ± 0.50 | 4.74 ± 1.18 * |
| Valerate | 0.0 ± 0.0 | 1.09 ± 0.55 * |
| Gene Symbol | Gene Name | Fold-Change (Mean BF/Mean FF) | q-Value |
|---|---|---|---|
| ARHGAP26 | Rho GTPase Activating Protein 26 | 4.96 | 0.100 |
| GPD2 | Glycerol-3-Phosphate Dehydrogenase 2 | 4.69 | 0.037 |
| NR3C1 * | Nuclear Receptor Subfamily 3, Group C, Member 1 | 4.65 | 0.039 |
| DEFB118 * | Defensin Beta 118 | 3.62 | 0.018 |
| PRKRA | Protein Activator of IFN Induced Protein Kinase | 3.62 | 0.087 |
| LTBP4 * | Latent TGF-ß Binding Protein 4 | 2.58 | 0.065 |
| CTNND1 * | Catenin Delta 1 | 2.54 | 0.065 |
| ARHGAP23 | Rho GTPase Activating Protein 26 | 2.31 | 0.091 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, K.; Donovan, S.M.; Ivanov, I.V.; Goldsby, J.S.; Davidson, L.A.; Chapkin, R.S. Assessing the Multivariate Relationship between the Human Infant Intestinal Exfoliated Cell Transcriptome (Exfoliome) and Microbiome in Response to Diet. Microorganisms 2020, 8, 2032. https://doi.org/10.3390/microorganisms8122032
He K, Donovan SM, Ivanov IV, Goldsby JS, Davidson LA, Chapkin RS. Assessing the Multivariate Relationship between the Human Infant Intestinal Exfoliated Cell Transcriptome (Exfoliome) and Microbiome in Response to Diet. Microorganisms. 2020; 8(12):2032. https://doi.org/10.3390/microorganisms8122032
Chicago/Turabian StyleHe, Kejun, Sharon M. Donovan, Ivan V. Ivanov, Jennifer S. Goldsby, Laurie A. Davidson, and Robert S. Chapkin. 2020. "Assessing the Multivariate Relationship between the Human Infant Intestinal Exfoliated Cell Transcriptome (Exfoliome) and Microbiome in Response to Diet" Microorganisms 8, no. 12: 2032. https://doi.org/10.3390/microorganisms8122032
APA StyleHe, K., Donovan, S. M., Ivanov, I. V., Goldsby, J. S., Davidson, L. A., & Chapkin, R. S. (2020). Assessing the Multivariate Relationship between the Human Infant Intestinal Exfoliated Cell Transcriptome (Exfoliome) and Microbiome in Response to Diet. Microorganisms, 8(12), 2032. https://doi.org/10.3390/microorganisms8122032