New Molecular Approach for the Detection of Kinetoplastida Parasites of Medical and Veterinary Interest


Abstract
:1. Introduction
2. Material and Methods
2.1. Primers and Probe’s Designs
2.1.1. Custom Protocol and In Silico Validation
2.1.2. Specificity-Based Principles of Oligonucleotide Design
2.2. Run Protocols
2.3. Conventional PCR Primer Sets Design, Amplification Protocol and Sequencing
2.4. PCR Systems Validation, Specificity, Sensitivity and Efficiency
2.5. PCR Tools Validation by Sample Screening and Identification of Kinetoplastida on Biological Samples
2.6. Determination of Assay Performance Characteristics
2.7. Statistical Analysis
3. Results
3.1. In Silico and In Vitro Validation
3.2. Determining Assay Performance Characteristics: Analytical Sensitivity, Linearity and Reproducibility
3.3. Performance Characteristics Comparison of the Diagnostic Tools
4. Discussion
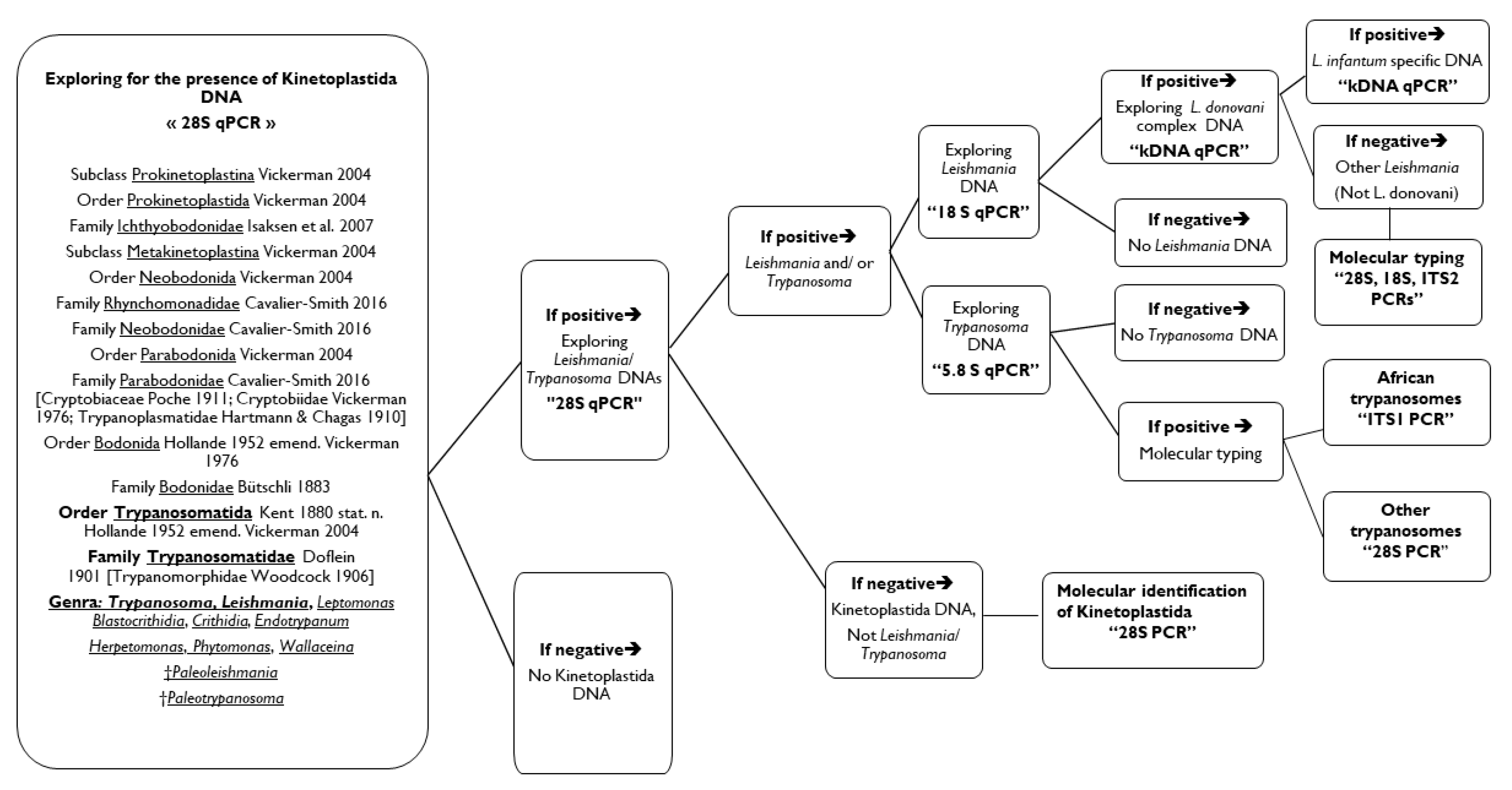
- For the detection of (practically) all Kinetoplastida parasites an initial screening using at least three qPCRs targeting three different genes for Kinetoplastida, i.e., screening with 28S Kinetoplastida qPCR with or without screening by the 28S pan-Leishmania/Trypanosoma, followed by screening with the 5.8S pan-Trypanosoma and the 18S pan-Leishmania qPCR assays.
- Trypanosoma spp. and Leishmania spp. qPCR assays, in addition to identifying Kinetoplastida at the genus level, they can decrypt co-infections Leishmania/Trypanosoma, allow parasite quantification and to define the therapeutic protocol and monitoring (molecules and doses).
- For the species identification, the 28S based PCR was able to identify Kinetoplastida, but at the genus or subgenus level. In addition, when there were infections by more than one species, it was not possible to sequence both amplicons.
- We can resolve this problem using genus-specific PCR systems, as was the case of co-infections by T. congolense/ L. infantum in two dogs from Cote d’Ivoire (Table S3). The kDNA based qPCRs identified L. infantum and L. donovani species without sequencing step.
5. Conclusions
Ethics approval and consent to participate
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavalier-smith, T. Higher classification and phylogeny of Euglenozoa. Eur. J. Protistol. 2016, 56, 250–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solter, L.F.; Becnel, J.J.; Vávra, J. Research methods for entomopathogenic microsporidia and other protists. Second Edi. In Manual of Techniques in Invertebrate Pathology; Elsevier Ltd.: Amsterdam, The Netherlands, 2012. [Google Scholar] [CrossRef]
- Simpson, A.G.B.; Stevens, J.R.; Lukes, J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006, 22, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Podlipaev, S. The more insect trypanosomatids under study-the more diverse Trypanosomatidae appears. Int. J. Parasitol. 2001, 31, 648–652. [Google Scholar] [CrossRef]
- Yazaki, E.; Ishikawa, S.A.; Kume, K.; Kumagai, A.; Kamaishi, T.; Tanifuji, G.; Hashimoto, T.; Inagaki, Y. Global Kinetoplastea phylogeny inferred from a large-scale multigene alignment including parasitic species for better understanding transitions from a free-living to a parasitic lifestyle. Genes Genet. Syst. 2017, 92, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overath, P.; Haag, J.; Lischke, A.; Colm, O. The surface structure of trypanosomes in relation to their molecular phylogeny. Int. J. Parasitol. 2001, 31, 468–471. [Google Scholar] [CrossRef]
- Kaufer, A.; Ellis, J.; Stark, D.; Barratt, J. The evolution of trypanosomatid taxonomy. Parasites Vectors 2017, 1–17. [Google Scholar] [CrossRef]
- Hemmige, V.; Tanowitz, H.; Sethi, A. Trypanosoma cruzi infection: A review with emphasis on cutaneous manifestations. Int. J. Dermatol. 2012, 51, 501–508. [Google Scholar] [CrossRef]
- Nussbaum, K.; Honek, J.; Cadmus, C.M.; Efferth, T. Trypanosomatid Parasites Causing Neglected Diseases. Curr. Med. Chem. 2010, 17, 1594–1617. [Google Scholar] [CrossRef]
- Rassi, A., Jr.; Marcondes de Rezende, J. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2012, 26, 275–291. [Google Scholar] [CrossRef]
- Alemayehu, B.; Alemayehu, M. Leishmaniasis: A Review on Parasite, Vector and Reservoir Host. Heal Sci. J. 2017, 11, 1–6. [Google Scholar] [CrossRef]
- Baldwin, T.M.; Elso, C.; Curtis, J.; Buckingham, L.; Handman, E. The Site of Leishmania major Infection Determines Disease Severity and Immune Responses. Infect. Immun. 2003, 71, 6830–6834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, L.J. Parasite-driven pathogenesis in Trypanosoma brucei infections. Parasite Immunol. 2011, 33, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [CrossRef] [PubMed]
- De Tommasi, A.S.; Otranto, D.; Dantas-Torres, F.; Capelli, G.; Breitschwerdt, E.B.; De Caprariis, D. Are vector-borne pathogen co-infections complicating the clinical presentation in dogs? Parasit. Vectors. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Eloy, L.; Lucheis, S. Canine trypanosomiasis: Etiology of infection and implications for public health. J. Venom. Anim. Toxins. Incl. Trop Dis. 2009, 15, 589–611. [Google Scholar] [CrossRef] [Green Version]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gürtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-cortés, A.; Ojeda, A.; Francino, O.; López-fuertes, L.; Timón, M.; Alberola, J. Leishmania Infection: Laboratory Diagnosing in the Absence of a “Gold Standard”. Am. J. Trop Med. Hyg. 2010, 82, 251–256. [Google Scholar] [CrossRef] [Green Version]
- De Paiva-Cavalcanti, M.; de Morais, R.C.; Pessoa-e-Silva, R.; Trajano-Silva, L.A.; Gonçalves-de-Albuquerque Sda, C.; Tavares Dde, H.; Brelaz-de-Castro, M.C.; Silva Rde, F.; Pereira, V.R. Leishmaniases diagnosis: An update on the use of immunological and molecular tools. Cell Biosci. 2015, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R. Advancing Molecular Diagnostics for Trypanosomatid Parasites. J. Mol. Diagn. 2014, 16, 379–381. [Google Scholar] [CrossRef]
- Alzohary, A.M. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Apte, A.; Daniel, S. Information Panel PCR Primer Design; CSHL Press: Cold Spring Harbor, NY, USA, 2009; Volume 4, pp. 1–10. [Google Scholar] [CrossRef]
- Owczarzy, R.; Tataurov, A.V.; Wu, Y.; Manthey, J.A.; McQuisten, K.A.; Almabrazi, H.G.; Pedersen, K.F.; Lin, Y.; Garretson, J.; McEntaggart, N.O.; et al. IDT SciTools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 2008, 36, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer- BLAST: A tool to design target- specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medkour, H.; Laidoudi, Y.; Athias, E.; Bouam, A.; Dizoé, S.; Davous, B.; Mediannikov, O. Molecular and serological detection of animal and human vector-borne pathogens in the blood of dogs from Côte d’Ivoire. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, M.E.; Steurer, F.J.; Koru, O.; Herwaldt, B.L.; Pieniazek, N.J.; Da Silva, A.J. Identification of Leishmania spp. by molecular amplification and DNA sequencing analysis of a fragment of rRNA internal transcribed spacer 2. J. Clin. Microbiol. 2011, 49, 3143–3149. [Google Scholar] [CrossRef] [Green Version]
- Njiru, Z.K.; Constantine, C.C.; Guya, S.; Crowther, J.; Kiragu, J.M.; Thompson, R.C.A.; Dávila, A.M.R. The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol. Res. 2005, 95, 186–192. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S Ribosomal DNA Amplification for Phylogenetic Study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [Green Version]
- Adl, S.M.; Simpson, A.G.; Lane, C.E.; David Bass, J.L.; Bowser, S.S.; Brown, M.; Burki, F.; Dunthorn, M.; Hampl, V.; Heiss, A.; et al. The Revised Classification of Eukaryotes. J. Eukaryot Microbiol. 2012, 59, 429–493. [Google Scholar] [CrossRef] [Green Version]
- Eickbush, T.H.; Eickbush, D.G. Finely orchestrated movements: Evolution of the ribosomal RNA genes. Genetics 2007, 175, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Turowski, T.W.; Tollervey, D. Transcription by RNA polymerase III: Insights into mechanism and regulation. Biochem. Soc. Trans. 2016, 44, 1367–1375. [Google Scholar] [CrossRef]
- Banda, M.; Bommineni, A.; Thomas, R.A.; Luckinbill, L.S.; Tucker, J.D. Evaluation and validation of housekeeping genes in response to ionizing radiation and chemical exposure for normalizing RNA expression in real-time PCR. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2008, 649, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Rebouças, E.D.L.; Costa, J.J.D.N.; Passos, M.J.; Passos, J.R.D.S.; Hurk, R.V.D.; Silva, J.R.V. Real time PCR and importance of housekeepings genes for normalization and quantification of mRNA expression in different tissues. Brazilian Arch. Biol. Technol. 2013, 56, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, M.; Galluzzi, L.; Migliazzo, A.; Magnani, M. Detection and Characterization of Leishmania (Leishmania) and Leishmania (Viannia) by SYBR Green- Based Real-Time PCR and High Resolution Melt Analysis Targeting Kinetoplast Minicircle DNA. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leutenegger, C.M.; Klein, D.; Hofmann-Lehmann, R.; Mislin, C.; Hummel, U.; Böni, J.; Boretti, F.S.; Günzburg, W.H.; Lutz, H. Rapid feline immunodeficiency virus provirus quantitation by polymerase chain reaction using the TaqMan® fluorogenic real-time detection system. J. Virol. Methods 1999, 78, 105–116. [Google Scholar] [CrossRef]
- Bustin, S.A. Quantitative PCR: A snapshot of current procedures and preferences. Expert Rev. Mol. Diagn. 2005, 30, 493–498. [Google Scholar] [CrossRef]
- Odiwuor, S.O.; Saad, A.A.; De Doncker, S.; Maes, I.; Laurent, T.; El Safi, S.; Mbuchi, M.; Büscher, P.; Dujardin, J.C.; Van der Auwera, G. Universal PCR assays for the differential detection of all Old World Leishmania species. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Medkour, H.; Davoust, B.; Levasseur, A.; Mediannikov, O. Molecular Evidence of Leishmania infantum and Leishmania guyanensis in Red Howler Monkey (Alouatta seniculus) from French Guiana. Vector-Borne Zoonotic Dis. 2019, 19, 1–5. [Google Scholar] [CrossRef]
- Medkour, H.; Laidoudi, Y.; Lafri, I.; Bitam, I.; Mediannikov, O.; Davoust, B. Canine leishmaniosis and first report of Leishmania infantum in the blood of equids in Kabylia (Algeria). Int. J. Infect. Dis. 2019, 79, 117–118. [Google Scholar] [CrossRef] [Green Version]
- Medkour, H.; Davoust, B.; Dulieu, F.; Maurizi, L.; Lamour, T.; Marié, J.-L.; Mediannikov, O. Potential animal reservoirs (dogs and bats) of human visceral leishmaniasis due to Leishmania infantum in French Guiana. PLoS Negl. Trop Dis. 2019, 13, e0007456. [Google Scholar] [CrossRef]
- Maslova, D.A.; Lukesb, J.; Jirkub, M.; Simpsorf, L. Phylogeny of trypanosomes as inferred from the small and large subunit rRNAs: Implications for the evolution of parasitism in the trypanosomatid protozoa. Mol. Biochem. Parasitol. 1996, 75, 197–205. [Google Scholar] [CrossRef]
- Hasan, G.; Turner, M.J.; Cordingley, J.S. Ribosomal RNA genes of Trypanosoma brucei: Mapping the regions specifying the six small ribosomal RNAs. Gene 1984, 27, 75–86. [Google Scholar] [CrossRef]
- Spencer, D.F.; Collings, J.C.; Schnare, M.N.; Gray, M.W. Multiple spacer sequences in the nuclear large subunit ribosomal RNA gene of Crithidia fasciculata. EMBO J. 1987, 6, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Downing, T.; Votýpka, J.; Kuhls, K.; Lukeš, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; Granouillac, B.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef]
- Vandersea, M.W.; Birkenheuer, A.J.; Litaker, R.W.; Vaden, S.L.; Renschler, J.S.; Gookin, J.L. Identification of Parabodo caudatus (class Kinetoplastea) in urine voided from a dog with hematuria. J. Vet. Diagn. Investig. 2015, 27, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Callahan, H.A. Molecular Taxonomy of the Suborder Bodonina (Order Kinetoplastida), Including the Important Fish Parasite, Ichthyobodo necator. J. Eukaryot. Microbiol. 2002, 49, 119–128. [Google Scholar] [CrossRef]
- Kunz, S. Learn more about Kinetoplastida. In Handbook of Cell Signaling, 2nd ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 2. [Google Scholar]
- Vexenat Ade, C.; Santana, J.M.; Teixeira, A.R. Cross-reactivity of antibodies in human infections by the kinetoplastid protozoa Trypanosoma cruzi, Leishmania chagasi and Leishmania (viannia) braziliensis. Rev. Inst. Med. Trop Sao Paulo. 1996, 38, 177–185. [Google Scholar] [CrossRef]
- Zanette, M.F.; Marçal, V.; Lima, F.D.; Laurenti, M.D.; Rossi, C.N.; Vides, J.P.; Vieira, R.F.; Biondo, A.W.; Marcondes, M. Serological cross-reactivity of Trypanosoma cruzi, Ehrlichia canis, Toxoplasma gondii, Neospora caninum and Babesia canis to Leishmania infantum chagasi tests in dogs. Rev. Soc. Bras Med. Trop. 2014, 47, 105–107. [Google Scholar] [CrossRef]
- Postigo, J.A. Leishmaniasis in the World Health Organization Eastern Mediterranean Region. Int. J. Antimicrob Agents. 2010, 36, S62–S65. [Google Scholar] [CrossRef]
- Lambson, B.; Smyth, A.; Barker, D.C. Leishmania donovani: Development and Characterisation of a Kinetoplast DNA Probe and Its Use in the Detection of Parasites. Exp. Parasitol. 2000, 94, 15–22. [Google Scholar] [CrossRef]
- Lachaud, L.; Marchergui-hammami, S.; Chabbert, E.; Dereure, J.; Dedet, J.P.; Bastien, P. Comparison of Six PCR Methods Using Peripheral Blood for Detection of Canine Visceral Leishmaniasis. J. Clin. Microbiol. 2002, 40, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Mary, C.; Faraut, F.; Lascombe, L.; Dumon, H. Quantification of Leishmania infantum DNA by a Real-Time PCR Assay with High Sensitivity. J. Clin. Microbiol. 2004, 42, 5249–5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
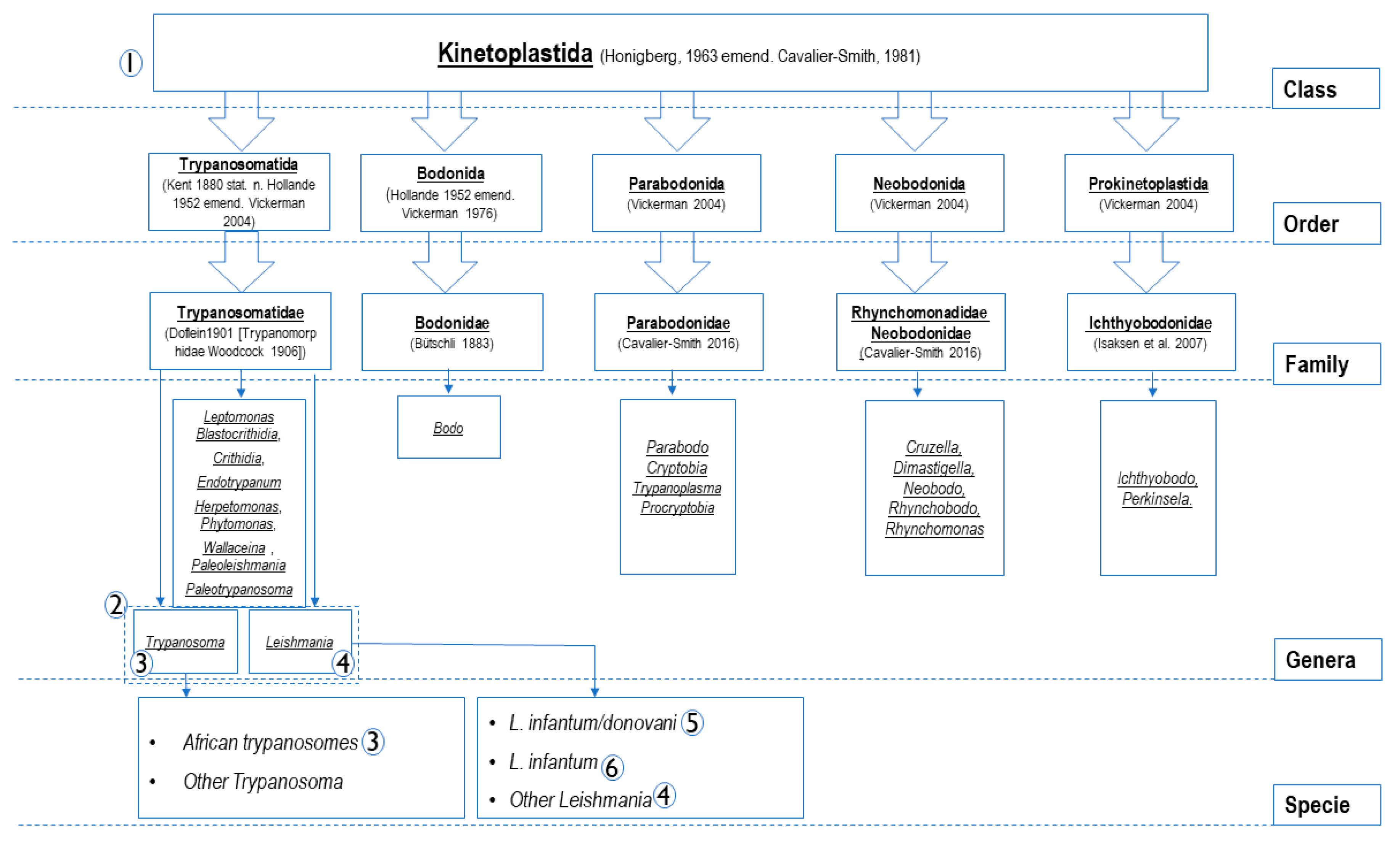
| PCR Name | Target Gene | Primers and Probes Name | Primers and Probes (5’-3’) | Amplicon Size (pb) | Tm °C | Specificity (Accordance to Figure 1) | Source |
|---|---|---|---|---|---|---|---|
| qPCR Pan-Kinetoplastida | 28S LSU (24 alpha) | P. 24a; 5345 | FAM-TAGGAAGACCGATAGCGAACAAGTAG | 200 | 60 °C | Kinetoplastida (1) | This study |
| F. 24a; 5198 | AGTATTGAGCCAAAGAAGG | ||||||
| R. 24a; 5412 | TTGTCACGACTTCAGGTTCTAT | ||||||
| qPCR Pan-Leishmania/ Trypanosoma | 28S LSU | P Leish/Tryp | FAM- GGGAAGGATTTCGTGCCAACG | 135 | 60 °C | Leishmania spp. and Trypanosoma spp. (2) | |
| F Leish/Tryp | AGATCTTGGTTGGCGTAG | ||||||
| R Leish/Tryp | ATAACGTTGTGCTCAGTTTCC | ||||||
| qPCR Pan-Trypanosoma | 5. 8 S rRNA | P. 5.8S Tryp | FAM-GTTGAAGAACGCAGCAAAGGCGAT | 83 | 60 °C | Trypanosoma spp. (3) | [25] |
| F. 5.8S Tryp | CAACGTGTCGCGATGGATGA | ||||||
| R. 5.8S Tryp- | ATTCTGCAATTGATACCACTTATC | ||||||
| qPCR Pan-Leishmania | 18S SSU | P. Leish | FAM- CGGCCGTAACGCCTTTTCAACTCA | 75 | 60 °C | Leishmania spp. (4) | |
| F. Leish | GGTTTAGTGCGTCCGGTG | ||||||
| R. Leish | ACGCCCCAGTACGTTCTCC | ||||||
| qPCR L. donovani/ L. infantum | kDNA minicircle | P. L. inf | FAM-TGGGCTGGATTGGGTTTTCCTGGGCTGGA | 175 | 60 °C | -VIC: L. donovani complex (5) -FAM: L. infantum (6) | This study |
| P. L. do cplx | VIC-TGGGCTCCCCTGGGCTGGATTGGGCTCC | ||||||
| F. L. inf/do | GGGGTTGGTGTAAAATAGGGCCGGGTGGT | ||||||
| R. L. inf/do | CCACATCAAAGGCACCCGAACCATTAA | ||||||
| PCR Pan-Kinetoplastida | 28S LSU | F2 | ACCAAGGAGTCAAACAGACG | (F0/R2) ~1300 (F2/R2) ~920 (F2/R1) ~550 | 53 °C 53 °C 58 °C | Possibility of hemi-nested PCR: F0/R2 than F2/R2. F2/R1 in direct PCR or hemi-nested after first amplification using F2/R2. Primers F0/R0 amplify ~800 pb at 53 °C with possibility of hemi-nested after amplification by F0/R2. F2/R1 primers more recommended. (1) | |
| R1 | GACGCCACATATCCCTAAG | ||||||
| R2 | GTTGGCACGAAATCCTTCC | ||||||
| F1 | ACCTAGTAGCTGGTTCAC | ||||||
| R0 | TCAGCATCGCTACAGGCCTC | ||||||
| PCR Pan-Leishmania | 18S SSU | F1 | CTGTGACTAAAGAAGCGTGAC | ~550 | 52 °C | Leishmania spp. (4) | |
| R1 | AGGCCGAATAGAAAAGATACGT | ||||||
| PCR Pan-Leishmania | ITS 2 | LGITSF2 | GCATGCCATATTCTCAGTGTC | 370 to 450 | 60 °C | Leishmania spp. (4) | [26] |
| LGITSR2 | GGCCAACGCGAAGTTGAATTC | ||||||
| PCR Pan-Trypanosoma | ITS 1 | ITS1-CF | CCGGAAGTTCACCGATATTG | 250 to 710 | 58 °C | African trypanosomes (3) | [27] |
| ITS1-BR | TTGCTGCGTTCTTCAACGAA |
| qPCR Assay DNA Targets | Kinetoplastida (28S) | Leishmania/Trypanosoma spp. (28S) | Trypanosoma spp. (5.8S) | Leishmania spp. (18S) | L. donovani/L. infantum (kDNA) | L. infantum (kDNA) |
|---|---|---|---|---|---|---|
| T. evansi Montecal EC8 | + | + | + | |||
| T. brucei gambiense biyiamina groupe II | + | + | + | |||
| T. brucei | + | + | + | |||
| T. brucei gambiense (T. Féo) | + | + | + | |||
| T. congolense (Chien Logan) | + | + | + | |||
| T. congolense IL 3000 | + | + | + | |||
| T. congolense (Dog) | + | + | + | |||
| T. cruzi CL Brunner | + | + | + | |||
| T. cruzi (Dog) | + | + | + | |||
| T. vivax | + | + | + | |||
| L. infantum | + | + | + | + | + | |
| L. donovani | + | + | + | + | ||
| L. major | + | + | + | |||
| L. guyanensis | + | + | + | |||
| Leptomonas sp. | + | |||||
| Bodo sp. | + | |||||
| L. infantum + L. donovani | + | + | ||||
| L. infantum + L. donovani + T. congolense IL 3000 | + | + | + | |||
| T. congolense IL 3000 + T. brucei | + | + | + | |||
| T. congolense IL 3000 + T. brucei + Leptomonas sp. | + | + | + | |||
| L.:Leishmania; T.:Trypanosoma |
| Coefficients of Variation Intra and Inter Assay for qPCR Assays | ||||||
|---|---|---|---|---|---|---|
| 28S Kinetoplastida spp. | 28S Leish/Trypano pp. | 5.8S Trypanosoma spp. | ||||
| T. Congolense Load | Intra Assay | Inter Assay | Intra Assay | Inter Assay | Intra Assay | Inter Assay |
| 1.06 × 106 | 1.61 | 2.84 | 5.13 | 1.10 | 1.87 | 4.43 |
| 1.06 × 105 | 1.88 | 2.15 | 4.24 | 3.04 | 5.28 | 7.71 |
| 1.06 × 104 | 2.90 | 1.82 | 1.70 | 0.37 | 3.44 | 7.60 |
| 1.06 × 103 | 1.78 | 9.25 | 2.72 | 3.40 | 2.63 | 8.80 |
| 1.06 × 102 | 0.85 | 0.68 | 2.47 | 2.66 | 1.11 | 4.80 |
| 1.06 × 101 | 3.38 | 6.97 | 1.76 | 1.42 | 1.99 | 1.61 |
| 1.06 × 100 | 3.61 | 9.06 | 2.12 | 2.89 | 0.17 | 2.05 |
| 1.06 × 10−1 | 1.54 | 9.87 | - | - | 2.22 | 3.12 |
| 1.06 × 10−2 | - | 1.94 | - | - | 0.24 | 1.25 |
| 1.06 × 10−3 | - | - | - | - | - | - |
| 18S Leishmania spp. | kDNA L. donovani cplx | |||||
| L. Donovani load | Intra Assay | Inter Assay | Intra Assay | Inter Assay | ||
| 1.00 × 104 | 1.19 | 1.03 | 0.94 | 2.55 | ||
| 1.00 × 103 | 1.54 | 0.42 | 1.05 | 0.99 | ||
| 1.00 × 102 | 0.32 | 0.21 | 0.54 | 2.90 | ||
| 1.00 × 101 | 0.22 | 2.00 | 0.41 | 1.81 | ||
| 1.00 × 100 | 1.45 | 5.74 | 0.26 | 0.72 | ||
| 1.00 × 10−1 | 0.97 | 1.16 | 1.47 | 0.37 | ||
| 1.00 × 10−2 | - | - | 0.73 | 0.59 | ||
| 1.00 × 10−3 | - | - | - | - | ||
| Statistic | TaqMan qPCR Systems | |||||
|---|---|---|---|---|---|---|
| 28S Kineto | 28S Leish-Tryp | 5.8S Tryp | 18S Leish | kDNA L. dono cplx | kDNA L. inf | |
| Correct classification | 0.962 | 0.965 | 0.986 | 0.984 | 0.997 | 0.998 |
| Misclassification | 0.038 | 0.035 | 0.014 | 0.016 | 0.003 | 0.002 |
| Sensitivity | 0.919 | 0.853 | 1.000 | 0.826 | 1.000 | 1.000 |
| Specificity | 0.967 | 0.976 | 0.986 | 0.994 | 0.997 | 0.997 |
| False positive rate | 0.033 | 0.024 | 0.014 | 0.006 | 0.003 | 0.003 |
| False negative rate | 0.081 | 0.147 | 0.000 | 0.174 | 0.000 | 0.000 |
| Prevalence | 0.100 | 0.092 | 0.035 | 0.062 | 0.060 | 0.051 |
| PPV (Positive predictive value) | 0.756 | 0.784 | 0.722 | 0.905 | 0.957 | 0.955 |
| NPV (Negative predictive value) | 0.991 | 0.985 | 1.000 | 0.989 | 1.000 | 1.000 |
| LR+ (Positive likelihood ratio) | 27.735 | 35.717 | 71.200 | 142.913 | 347.000 | 390.000 |
| LR− (Negative likelihood ratio) | 0.084 | 0.151 | 0.000 | 0.175 | 0.000 | 0.000 |
| Relative risk | 81.600 | 52.043 | - | 78.714 | - | - |
| Odds ratio | 330.727 | 237.075 | - | 817.000 | - | - |
| Cohen’s Kappa | 0.81 | 0.800 | 0.832 | 0.855 | 0.976 | 0.975 |
| Agreement* | almost perfect | substantial | almost perfect | almost perfect | almost perfect | almost perfect |
| TaqMan qPCR Target | Detected and Typed | Detected, Untyped | Typed, Not Detected | Not Detected, Untyped |
|---|---|---|---|---|
| 28S Kinetoplastida | 34 | 11 | 3 | 321 |
| 28S Leishmania/ Trypanosoma | 29 | 8 | 5 | 327 |
| 5.8S Trypanosoma spp. | 13 | 5 | 0 | 351 |
| 18S Leishmania spp. | 19 | 2 | 4 | 344 |
| kDNA L. donovani complex | 22 | 1 | 0 | 346 |
| kDNA L. infantum | 21 | 1 | 0 | 347 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medkour, H.; Varloud, M.; Davoust, B.; Mediannikov, O. New Molecular Approach for the Detection of Kinetoplastida Parasites of Medical and Veterinary Interest. Microorganisms 2020, 8, 356. https://doi.org/10.3390/microorganisms8030356
Medkour H, Varloud M, Davoust B, Mediannikov O. New Molecular Approach for the Detection of Kinetoplastida Parasites of Medical and Veterinary Interest. Microorganisms. 2020; 8(3):356. https://doi.org/10.3390/microorganisms8030356
Chicago/Turabian StyleMedkour, Hacène, Marie Varloud, Bernard Davoust, and Oleg Mediannikov. 2020. "New Molecular Approach for the Detection of Kinetoplastida Parasites of Medical and Veterinary Interest" Microorganisms 8, no. 3: 356. https://doi.org/10.3390/microorganisms8030356
APA StyleMedkour, H., Varloud, M., Davoust, B., & Mediannikov, O. (2020). New Molecular Approach for the Detection of Kinetoplastida Parasites of Medical and Veterinary Interest. Microorganisms, 8(3), 356. https://doi.org/10.3390/microorganisms8030356