4.1. Nature and Extent of C. trachomatis Plasmid Diversity
Advancements in genome sequencing technology over the past 20 years have shed light on many aspects of chlamydial biology and epidemiology, and with hundreds of whole genome sequences now in the public domain we have the ability to delve into the evolutionary history of
C. trachomatis to a depth not previously possible. We now understand that modern lineages are the product of thousands of years of evolution rather than millions [
49], and that the chlamydial plasmid has been vertically inherited throughout its evolutionary history, with very few instances of recombination or exchange between lineages [
16,
35,
49,
62]. Furthermore, through genetic manipulation and in vivo experiments, the role of each plasmid CDS in chlamydial virulence, regulation of gene expression and plasmid maintenance are gradually being revealed (
Table 1). However, few studies have performed in-depth analyses on multiple
C. trachomatis plasmid sequences [
16,
35,
62,
63], and most of these studies considered relatively few isolates (the largest study up until now included 157 sequences in their analysis [
62]. In the present study, we analysed 524
C. trachomatis plasmid sequences, providing the largest in-depth study on chlamydia plasmid diversity to date. Analysis of this larger dataset has resulted in an increased capture of
C. trachomatis diversity, with plasmid variation nearly three times greater (at 2.97%) than previously calculated [
16,
64]. The discovery of more variation seems an inevitable consequence of analysing increasingly large datasets due to the continual occurrence of point mutations. But, due to the relatively small availability of mutable sites in the plasmid (i.e., without incurring a fitness cost), we wondered if the large size of the current dataset and wide geographic and temporal distribution completes the picture of plasmid variation in
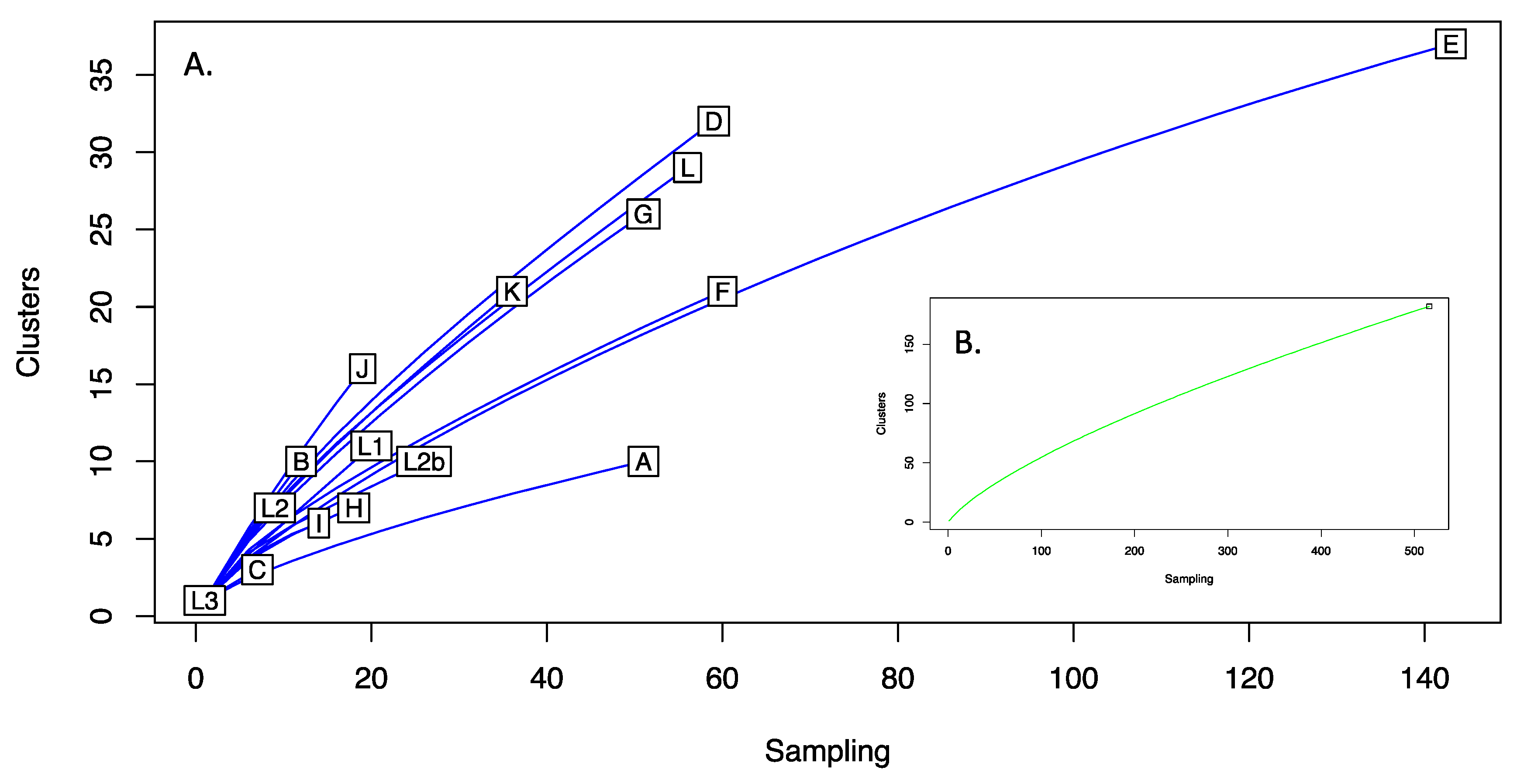
C. trachomatis. In fact, it appears that this is not the case. The rarefaction curves generated upon resampling of data for the entire species and each genotype in isolation suggested that hitherto undescribed variation likely exists among
C. trachomatis plasmids (
Figure 2), and therefore further sampling may be warranted. The current availability of sequences is inherently biased towards the more common genotypes (such as E) (
Figure 2), with some rarer types existing as sole surviving representatives (e.g., L3) or in very low numbers (e.g., C, I, H, L2). Therefore, the analysis of more examples of the rarer genotypes is likely to increase the diversity captured, highlighting the importance of collecting and preserving diverse isolates from divergent locations [
22].
The majority of variation currently described within the plasmid consists of SNPs, with large-scale deletions and recombination events both being relatively rare [
49] and indeed we did not identify any events such as these other than what has been previously described [
16,
35,
49]. Additionally, CDS length was highly conserved with only one premature stop codon identified and two delayed stop codons, each causing only minor changes to the length of the CDS. This conservation of CDS length highlights the importance of the plasmid to chlamydial survival.
Some diversity was identified within the replication origin of the plasmid, which comprises of (usually) four 22 bp repeat sequences [
16]. Almost half of the sequences analysed here had four identical 22 bp repeats, and a further 34% of sequences had three complete and a fourth incomplete repeat sequence. This supports the notion that the 22 bp repeat sequence is important to plasmid maintenance and suggests that at least three repeats are required for efficient replication without affecting copy number [
16]. However, in the present study a number of sequences had fewer repeats, with the third most common iteration being one perfect and one imperfect repeat (10%), particularly in E, F and L2b genotypes. However, the numbers are low, and it seems most likely that this is due to the high stringency of the assembly and alignment process rather than being a biological phenomenon; indeed, visual inspection of the read data showed that in the two isolates that apparently lacked a repeat region entirely, four repeat sequences were actually present. This observation was confirmed across five other randomly selected sequences that were reported as containing fewer than three repeats, and in each case, at least three repeat sequences were identified, highlighting the need to verify assembled genomes against read data.
Individual SNPs within coding sequences can have serious implications on the encoded protein due to alternations to the encoded amino acid sequence, and the position of the SNP within the codon is what determines its impact. The third base position of a codon is highly redundant, as around 67% of mutations at this locus are synonymous. The remaining 33% of mutations are nonsynonymous, but the physical characteristics of the encoded amino acid are maintained [
65] and so the effect on the resulting protein will be minimal. The first codon position will result in a synonymous mutation in only 4% of cases, and second position mutations are always nonsynonymous; furthermore, most substitutions will always result in a change in amino acid characteristics at these sites [
65]. As a result, one would expect the most common variable nucleotide position to be the third position, and indeed we found that across the entire plasmid 52.9% of intragenic SNP loci were at the third base position in the codon. However, CDS1 had an unusually high proportion of SNP loci in the first base position (51.6%), reflecting the previously identified redundancy of this gene in plasmid maintenance [
16,
17]. This is in stark contrast to its predicted functional homologue, CDS2, in which just 4% of SNP loci occurred at the first base position. Concordantly, CDS2 had the lowest percentage of nonsynonymous SNPs and can be considered the most functionally conserved gene of the plasmid, presumably due to its role in plasmid maintenance, confirming the much earlier study by Seth-Smith et al., (2009) [
16]. Whilst variation in the CDS2 amino acid sequence was found to be uniformly low, variation in the nucleic acid sequence was found to be relatively high (fourth highest among the eight CDS). This may be explained by the existence of the overlapping sRNA-2 sequence, for which variations in sequence may form part of its function. Many sRNA molecules are involved in binding of mRNA, affecting their stability and/or translation [
66]; others directly bind to protein transcription factors, affecting gene expression [
67]; furthermore, the presence of SNPs in sRNA may alter the affinity to targeted mRNA and thus could have a significant effect on gene regulation [
68]. Although the role of sRNA-2 is not yet understood, its expression levels at 12 h post infection [
8] suggests an important role for sRNA-2 in regulation of genes important midway through the developmental cycle, such as RB replication, or possibly the early stages of RB-EB conversion [
69,
70,
71].
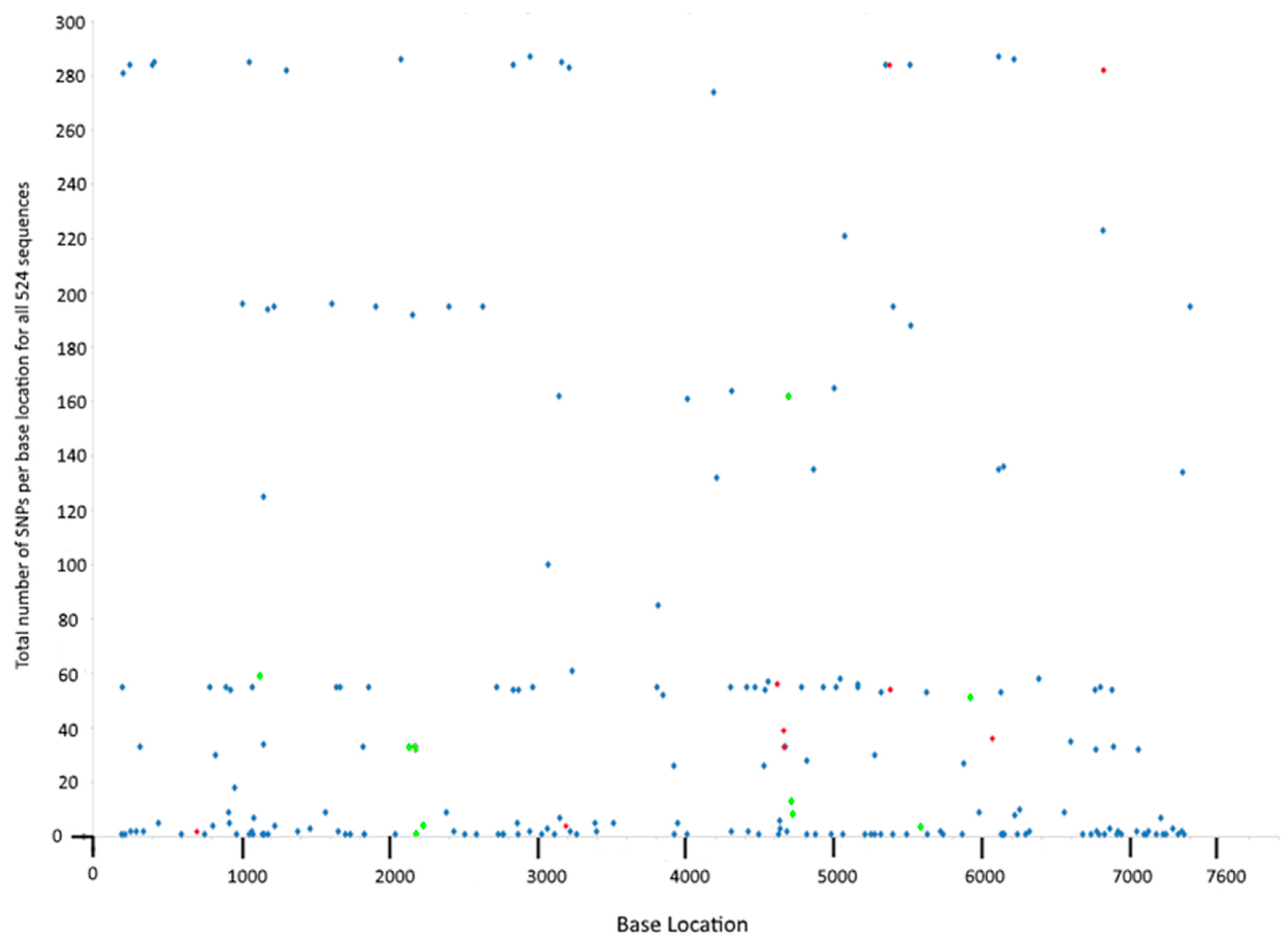
In this study, each isolate was sampled at a single time point so there is no way of knowing whether that particular SNP variant later expanded to become a dominant clone, or if it vanished from existence due to deleterious effects—although predictions can be made based on their frequency in the dataset. The latter outcome possibly befell the most infrequently sampled SNPs, particularly the 60 SNPs that occurred only once among all isolates included in the study (
Figure 1) which may represent transient events—although they may also indicate variation among isolates from under-sampled locations. The remaining SNPs were present in multiple sequences (
Figure 1 and
Table S2). The very frequently sampled SNPs were branch-specific (no homoplasic SNPs were identified in this dataset), occurring early in the evolution of
C. trachomatis. These became fixed either through chance (i.e., they are irrelevant to survival) or through offering a selective advantage to those strains carrying them. Tissue tropism-determining SNPs are well documented in the chlamydial chromosome (see reviews by [
72,
73,
74]) but few have been identified in the plasmid [
16]. Indeed, we did not identify any SNPs that consistently differentiated ocular from urogenital trachoma isolates; the SNP found to be unique to ocular strains by Seth-Smith et al. (2009) [
16] was also found to occur in sporadic genotype G (G_S4658 and G_Ar246) and J (J_UK583676, J_UK35672, J_Soton72 and J_S42) isolates in the present dataset (these sequences were not available at the time of the earlier study). This reflects the close relatedness between these and trachoma (serovar A) plasmids previously noted [
62], and may result from plasmid-swapping between strains, or recombination between plasmids; there is prior evidence of this from the present dataset [
49]. Also, there is precedence for recombination between ocular and urogenital isolates [
75] so the opportunity for genetic exchange must exist.
However, 30 SNPs were identified that consistently differentiated the LGV biovar from urogenital or ocular strains. Of these, 15 were synonymous mutations and so may have been retained through random genetic drift early in the evolutionary history of chlamydia, (i.e., at the point of divergence between trachoma and LGV lineages), and these mutations were then carried passively through subsequent evolutionary events. Possible exceptions are those synonymous mutations that fall within the two sRNA sequences overlapping CDS2 and CDS7, which may have an effect on secondary structure or mRNA binding. The 15 nonsynonymous mutations identified that divide LGV strains from the trachoma biovar are more likely to have arisen through natural selection due to the potential biological consequences of changes to amino acid sequences. Seven of these nonsynonymous SNPs occurred in CDS5 (
Table S5). The gene product of CDS5, Pgp3, is the only plasmid-encoded protein secreted into the inclusion lumen and cytosol [
23]. Pgp3 may be important in host cell invasion [
21], and has recently been identified as being an inhibitor of apoptosis in cell culture, via activation of the PI3K/AKT signalling pathway [
32]. This interaction of Pgp3 with host cell signalling pathways suggests that CDS5 may be subject to immune selection, and the accumulation of LGV-specific nonsynonymous SNPs in this gene may suggest a role in LGV tropism, a notion supported by the five-fold higher expression of Pgp-3 in LGV compared to ocular strains [
8]. Additionally, the crystal structures of Pgp3 from a urogenital (serovar D) and LGV (L1 440) strain have been resolved [
21,
22]. Pgp3 differs in structure between the biovars, with nine amino acid changes being identified between the two strains, resulting in the LGV version of Pgp3 occupying a different space group to that of the serovar D Pgp3 protein [
22]. Across the present dataset, seven of the nine previously identified LGV-specific changes are maintained, but two amino acid replacements (T39K and D86N) also occur in many ocular and urogenital trachoma strains so are unlikely to contribute to LGV tropism. The two amino acids marked as being functionally significant in receptor binding (phenylalanine at amino acid site 6, and tryptophan at site 234) [
22] are conserved across all sequences in the present dataset; however, a complete understanding of how the Pgp3 structure affects its biological function remains to be determined.
Evidence that these CDS5 mutations have become fixed in the LGV biovar due to natural selection has yet to be provided, although attempts at investigating signs of selection in the
C. trachomatis plasmid have been made using the dN/dS ratio [
8,
62]. Both studies found that although CDS5 had a dN/dS ratio indicative of positive selection (dN/dS > 1), this value was not statistically significant. As the present dataset is much larger than those previously analysed, we wondered whether the dN/dS ratio would tip towards statistical significance as a result of the additional sequences included in the analysis. But once again, none of the codons of any plasmid CDS, including CDS5, were found to significantly depart from neutrality. This is surprising given the near-ubiquitous carriage of the plasmid by clinical strains of
C. trachomatis and the conservation of most of the plasmid-borne genes among diverse isolates; it seems highly unlikely that this has occurred by chance. However, it should be acknowledged that when the dN/dS ratio was developed, it was not intended for within-species comparisons; rather, the test was developed to analyse representative sequences of divergent species, where mutations are considered to be fixed [
53]. When using this analysis for within-species data (where many mutations are transient in nature), this underlying assumption is violated [
76]. As a result, the test is too conservative and results may be misleading [
76,
77]. Further work is needed before firm conclusions can be drawn about the effect of selective pressure on
Chlamydia plasmid evolution.
4.2. Implications on Diagnostic Target Choice
Modern diagnostic approaches are mainly based on nucleic acid amplification techniques, as these are highly sensitive and specific to the targeted organism. However, a complete understanding of the variation at the chosen target sites in the genome is essential for the continued efficacy of the test. In addition to the case of the Swedish New Variant, where a 344 bp deletion in the plasmid resulted in elimination of the single target site of a major diagnostic assay [
78], there is now a second example where mutation of a single diagnostic target site has led to large-scale false-negative reporting of
C. trachomatis [
79]. The Finnish New Variant escaped detection by the Aptima Combo-2 test, which targets the 23S rRNA, but remained detectable by the Aptima CT test, which targets a sequence within the 16S rRNA sequence. The global distribution of the Finnish New Variant has yet to be determined, but it has also been detected in Sweden and further reports seem likely [
80]. It is not known whether these isolates were imported from Finland or represent a separate evolutionary event—the former seems more likely given the close proximity of the two countries, but the latter cannot be ruled out until further investigations are completed. These examples of diagnostic failure highlight the importance of building a thorough understanding of target stability prior to employing a particular diagnostic target, with a focus on future stability being of paramount importance. Plasmid DNA tends to be present in multiple copies, thus targeting sequences within the plasmid affords a greater sensitivity of detection than chromosomal sites. However, the plasmid is accessory to survival; plasmid-free
C. trachomatis isolates have been detected in clinical samples, although they are extremely rare [
37,
43,
44,
81,
82]. It has previously been suggested that the plasmid might be a poor diagnostic target due to the opportunity for homologous recombination between plasmids and exchange of plasmids between isolates [
35]; however, these events are infrequent. Accordingly, diagnostic tests employing dual-target assays that target both chromosomal and plasmid sequences should be preferentially considered to mitigate the risk of target deletion or plasmid loss, whilst retaining high sensitivity for low-level infections [
83,
84,
85].
The relative stabilities of potential plasmid-based diagnostic targets were assessed by analysing the degree of sequence diversity at those sites. The first study to analyse multiple plasmid sequences identified CDS2 as being the most conserved gene at the nucleotide level [
16]. However, more recently, CDS6 was identified as being the most highly conserved plasmid gene [
62], and results from the present study support the latter study. Firstly, the SNP rate was determined by simply comparing each CDS on the number of SNP loci, normalized by dividing this by the total number of nucleotides in the gene. We found that CDS8 had the highest SNP rate (3.9%) whereas CDS6 had the lowest (1.94%), with CDS2 falling roughly in the middle, when considering only the number of SNP loci within the gene. A SNP that occurs frequently in the dataset is not necessarily more informative on evolutionary processes than a SNP that occurs infrequently, as a frequent SNP may be present in an over-represented genotype or may have occurred early in the evolutionary history of
C. trachomatis with no effect on fitness. Such a SNP may be passively carried through subsequent generations. Nonetheless, this type of site may be informative for diagnostic target selection, as if it does not impair fitness it may have more chance of reverting to the ancestral state, which may result in reduced affinity between target site and diagnostic probe. In fact, the calculation of genetic distance (which considers the number of sequences carrying a SNP at each locus) found that whilst CDS6 retained the lowest value (d = 0.001), whereas the most variable gene was CDS5 (d = 0.007). Taken together, this suggests CDS6 may be a good choice for selection of diagnostic target sites. However, CDS6 is nonessential for stable plasmid maintenance in tissue culture [
46], and its necessity in vivo has not yet been assessed. Nonetheless, CDS6 encodes the pgp4 protein, which has a role in the plasmid’s ability to accumulate glycogen [
46] and is the sole regulator of pgp3 [
19,
86] and other virulence associated genes, suggesting CDS6 is important in the context of the natural host. Along with the presence of only 6 variable sites throughout the gene and the relative rarity of these SNPs in the dataset, this suggests that selective pressure exerted by diagnostic detection will be overcome by natural selection, resulting in its continued low variability. A disadvantage of using this gene would be the relative shortness of the coding sequence at only 309 bp in length, which may present challenges in optimizing primer design. A compromise between pragmatism and sequence stability needs to be reached when choosing optimal diagnostic target sites.
A potential alternative diagnostic target to CDS6 may be offered by CDS3. Although CDS3 had the highest number of SNP locations, it had the second lowest SNP rate, once the length of the CDS has been taken into account (
Table 2). CDS3 is the longest of the plasmid CDS features, which provides more options for the design of optimal primer pairs. Furthermore, the function of CDS3 has been assigned for some time and it is known to be essential for stable plasmid maintenance [
19]. Thus, CDS3 may provide a useful alternative for assay design, if conserved sequences within CDS3 are targeted. This finding reflects the change implemented by Abbott Laboratories, who introduced the Abbott RealTime CT/NG assay, a dual plasmid assay combining primers for CDS1 and CDS3, replacing the single target Abbott m2000 assay which failed to detect the nvCT strain [
87]. However, the continued use of CDS1 as a diagnostic target site is questionable given the high degree of variation seen in this gene in the present study and others [
8,
16,
35,
49,
62].
The presence of 30 biovar-specific SNPs identified in this study may be useful for diagnostic purposes and could aid in identifying LGV infections in the clinic. This is important in the choice of treatment regimens, which differ due to a delayed antibiotic cure rate of LGV when compared to urogenital chlamydia infection [
88,
89]. Melt curve analyses using probes that target LGV-specific SNPs have been designed to discriminate LGV infections from urogenital strains, based on SNPs within the
ompA and
pmpH genes [
90,
91], but with the increased sensitivity afforded by the multi-copy plasmid, this information could offer a useful alternative.

 ,
, 

{kind=link}
{kind=link}