Gut Microbial Protein Expression in Response to Dietary Patterns in a Controlled Feeding Study: A Metaproteomic Approach


 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants
2.3. Study Diets
2.4. Sample Collection and Processing
2.5. Protein Extraction and Sample Preparation
2.6. LC MS/MS Analysis
2.7. Database Search
2.8. Human Protein Data Analysis
2.9. Metaproteomic Data Analysis
2.10. Bioinformatics and Statistical Analysis
3. Results
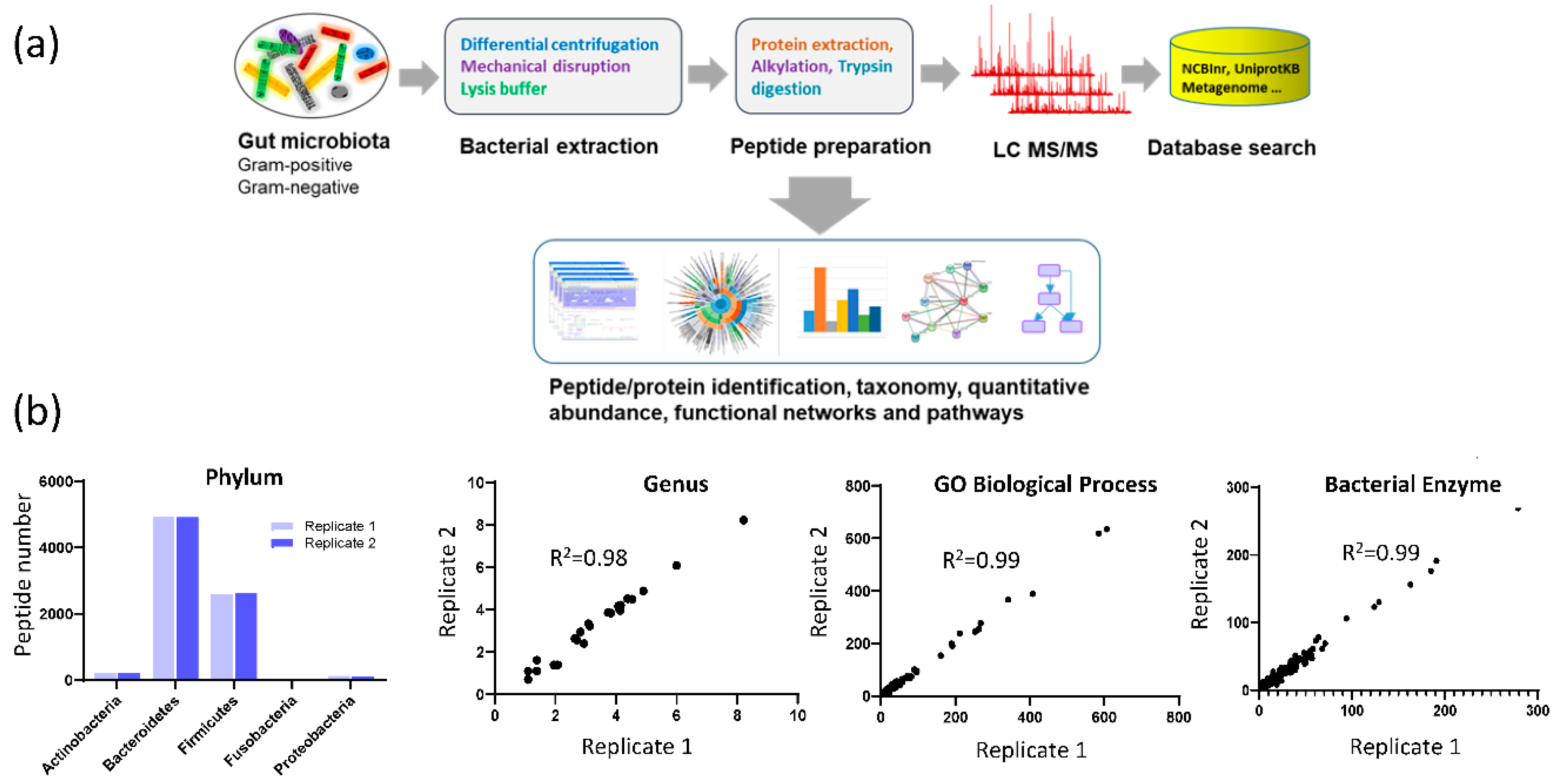
3.1. Assay Reproducibility
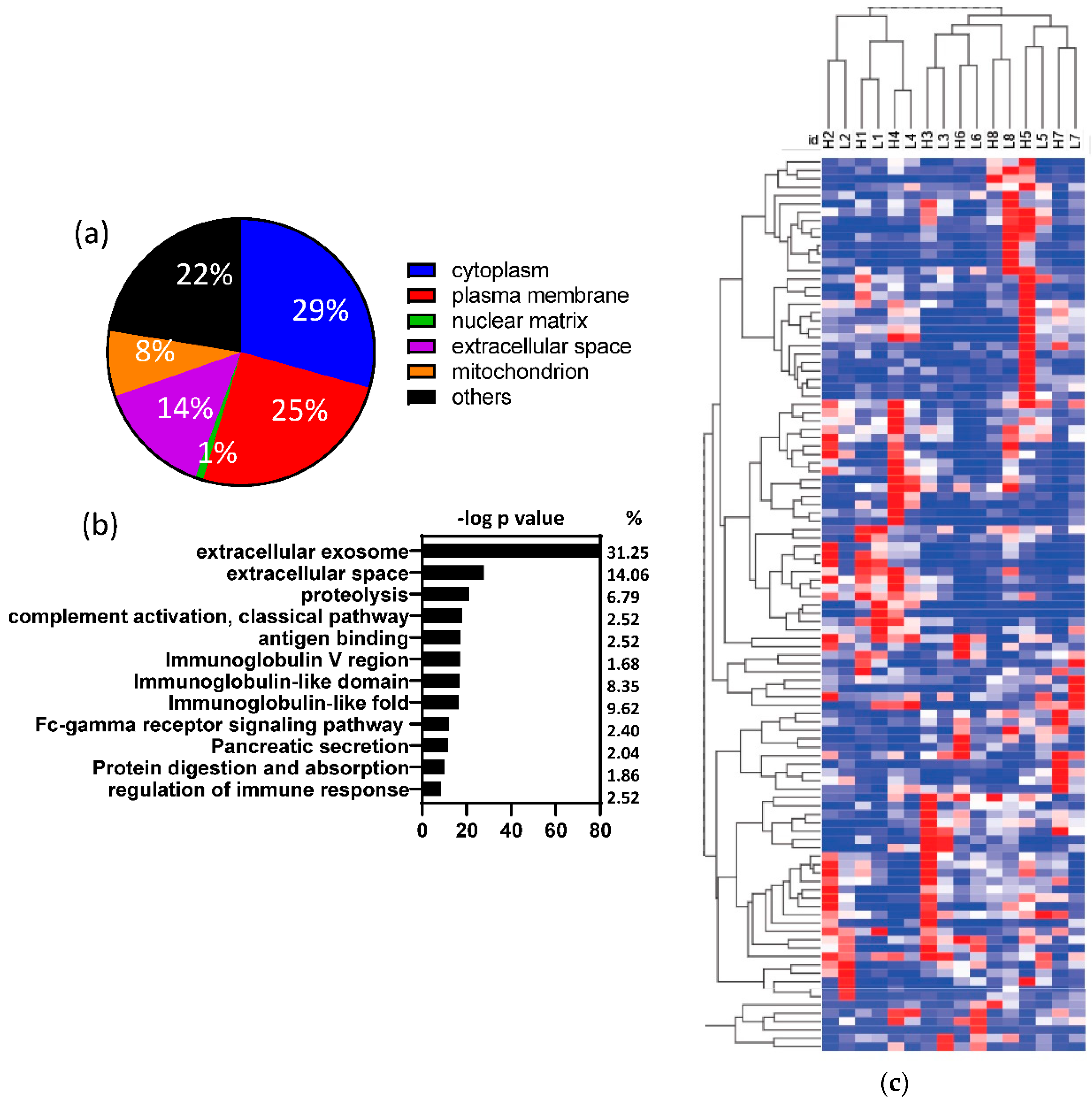
3.2. Human proteins identified in fecal samples
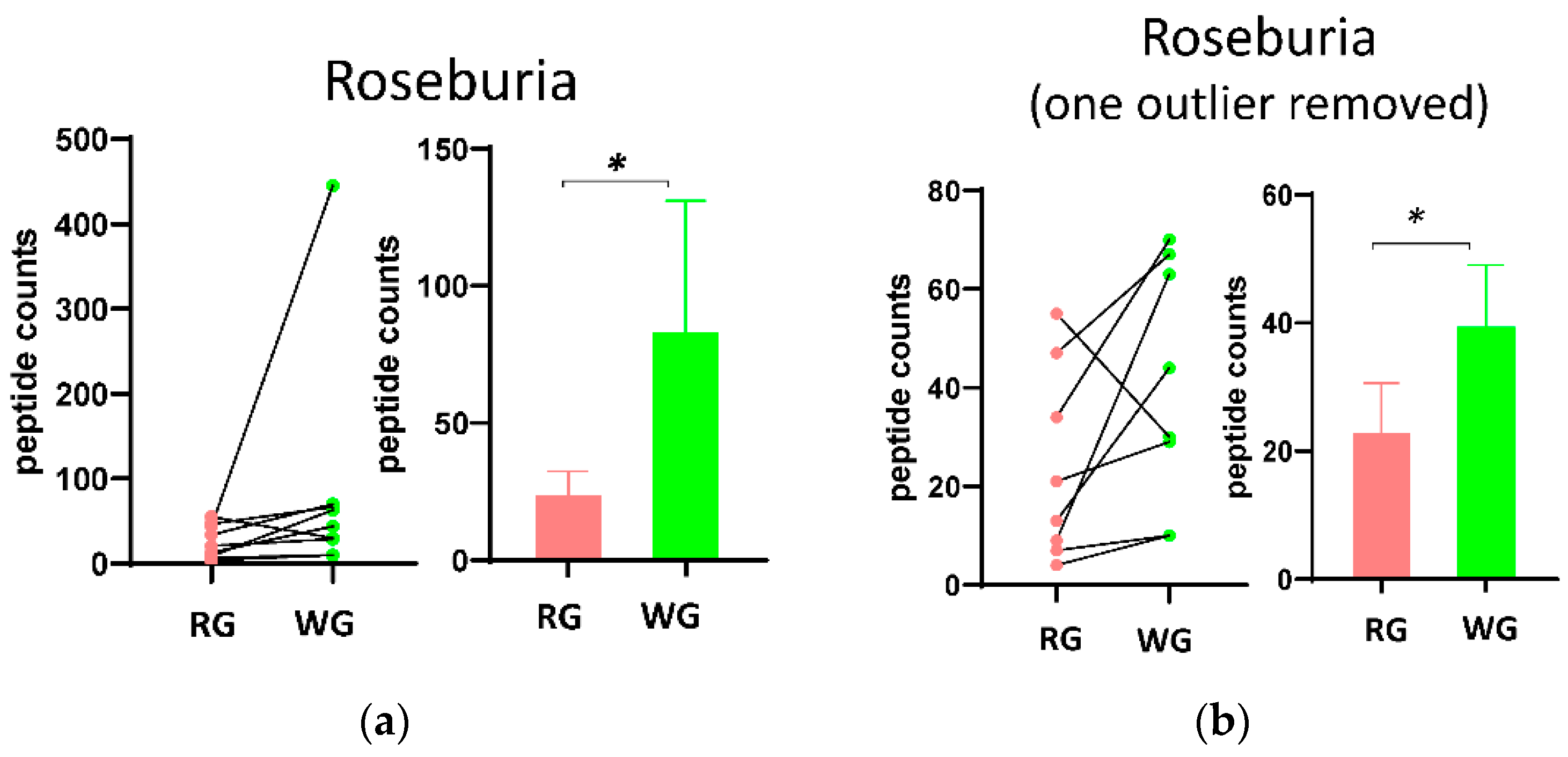
3.3. Fecal Microbial Composition Based on Metaproteomics Analysis
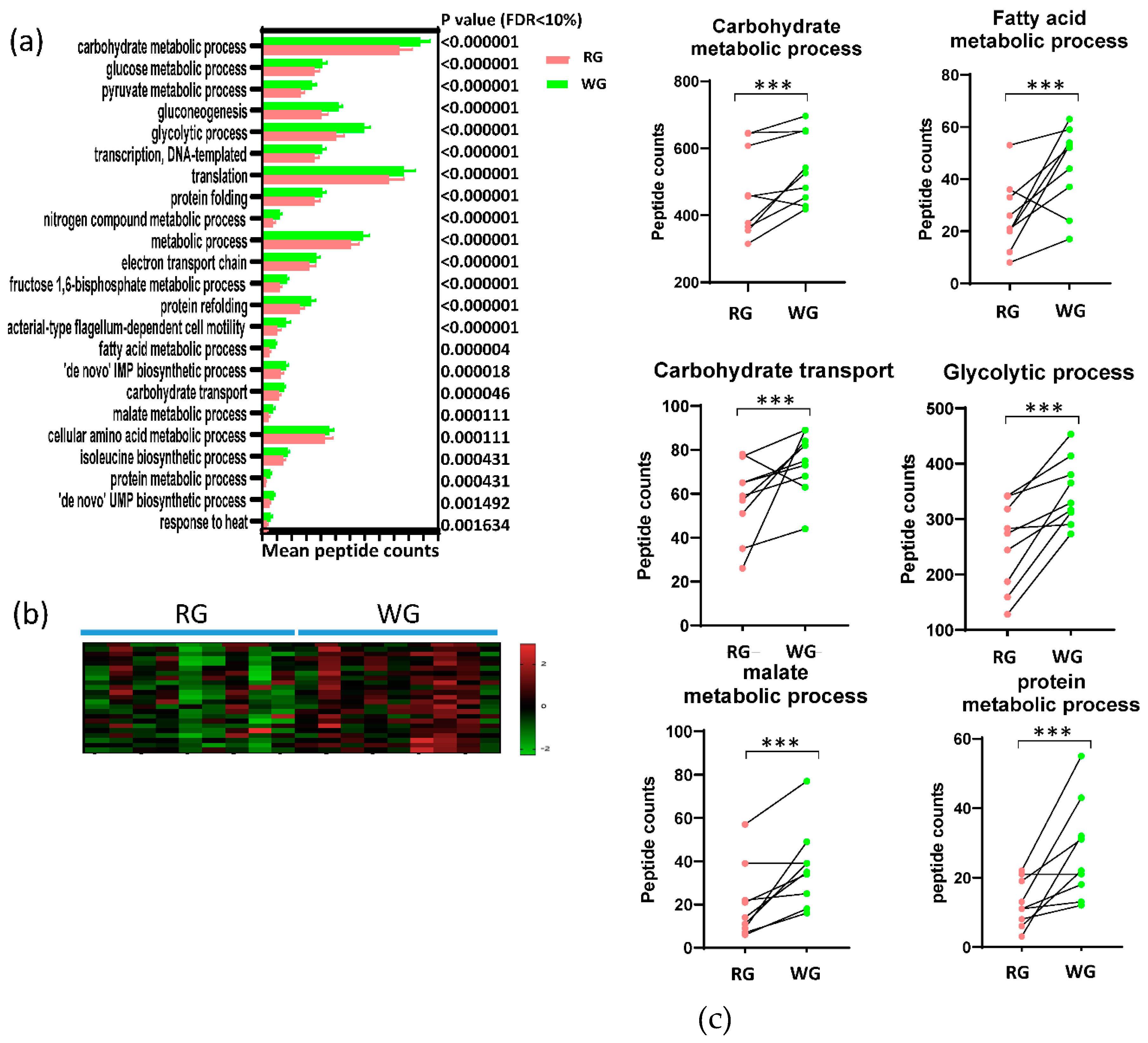
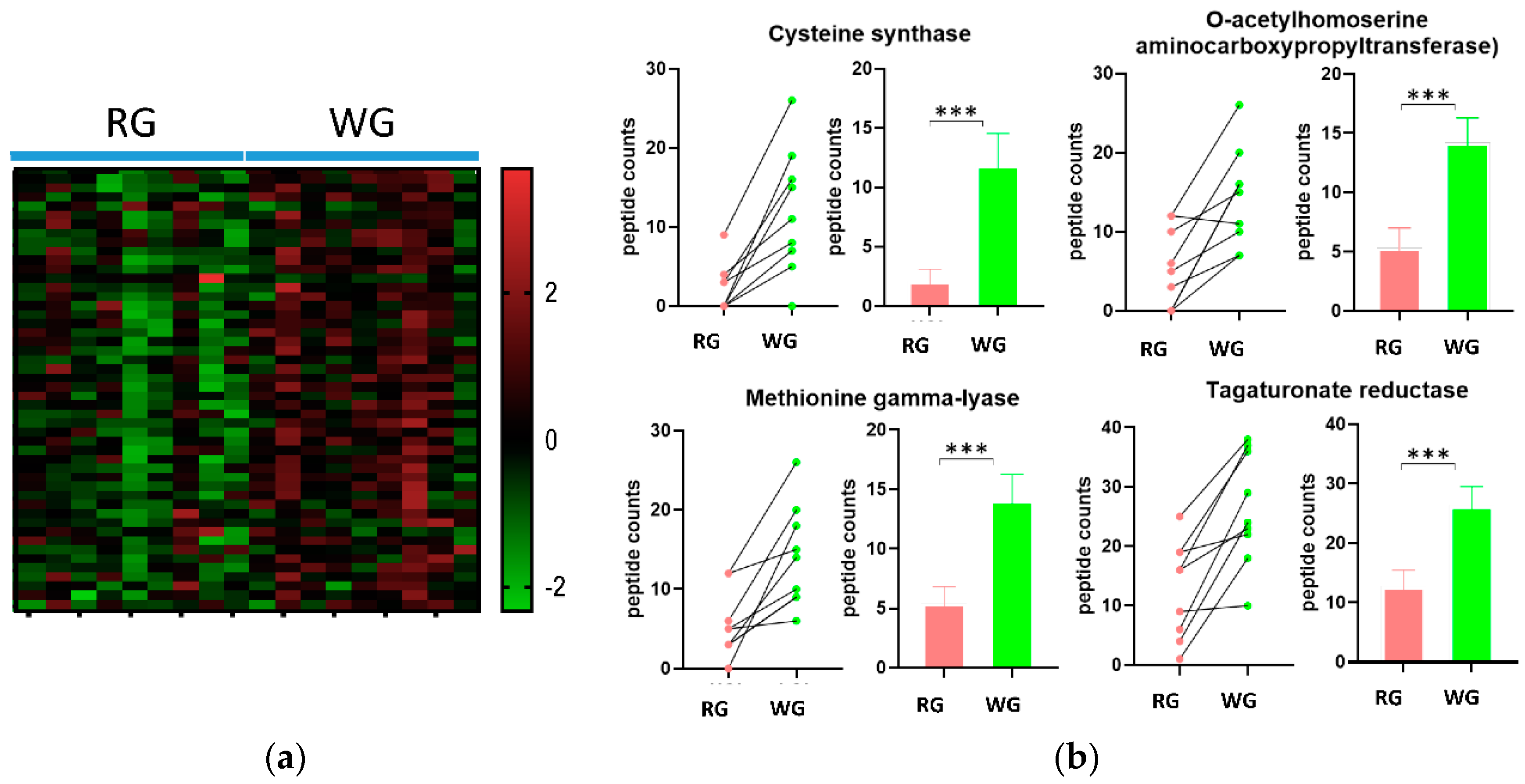
3.4. Diet-Induced Significant Changes in GO Biological Processes
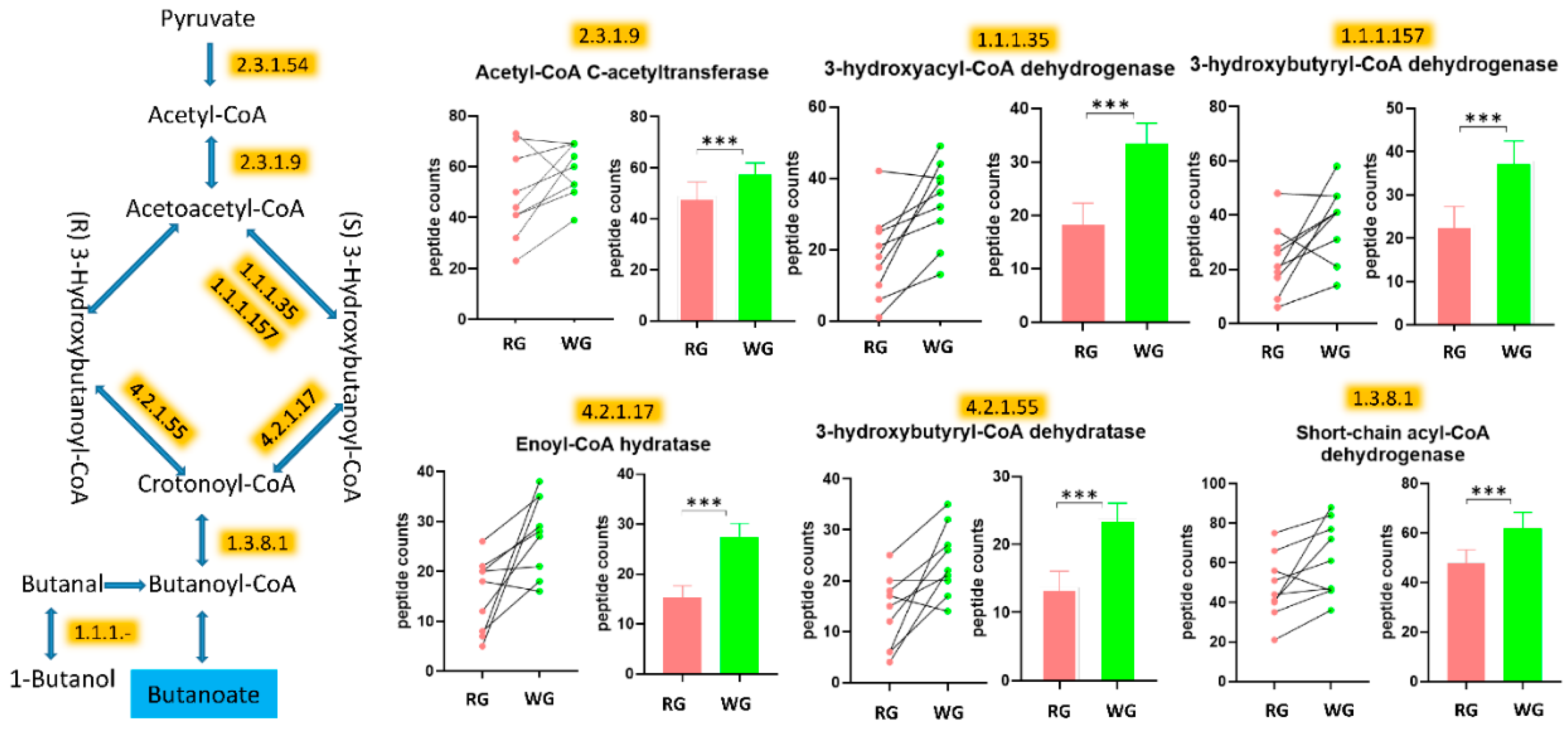
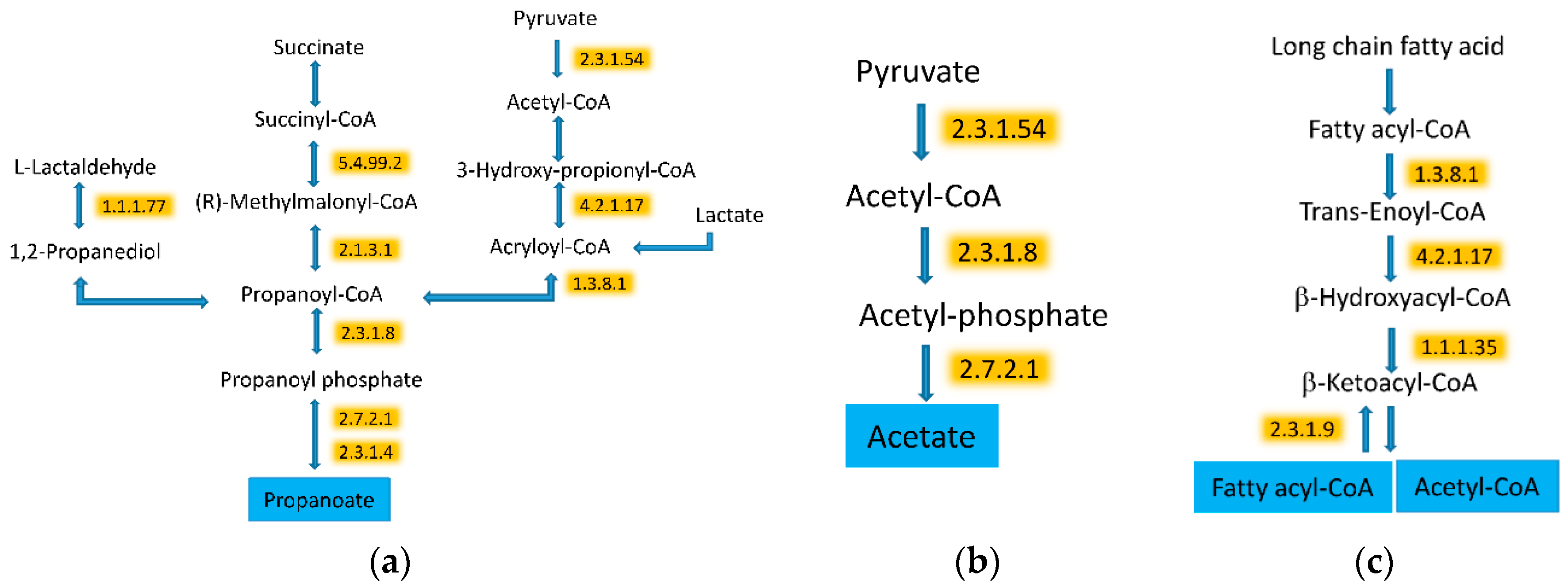
3.5. WG Induced Significant Enrichment in Bacterial Enzymes Involved in the Production of SCFA and the Degradation of Fatty Acids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maier, T.V.; Lucio, M.; Lee, L.H.; VerBerkmoes, N.C.; Brislawn, C.J.; Bernhardt, J.; Lamendella, R.; McDermott, J.E.; Bergeron, N.; Heinzmann, S.S.; et al. Impact of dietary resistant starch on the human gut microbiome, metaproteome, and metabolome. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Walker, A.; Haange, S.B.; Lagkouvardos, I.; Liu, Y.; Schmitt-Kopplin, P.; von Bergen, M.; Jehmlich, N.; He, X.; Clavel, T.; et al. Synbiotic-driven improvement of metabolic disturbances is associated with changes in the gut microbiome in diet-induced obese mice. Mol. Metab. 2019, 22, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Neuhouser, M.L.; Schwarz, Y.; Wang, C.; Breymeyer, K.; Coronado, G.; Wang, C.Y.; Noar, K.; Song, X.; Lampe, J.W. A low-glycemic load diet reduces serum C-reactive protein and modestly increases adiponectin in overweight and obese adults. J. Nutr. 2012, 142, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Runchey, S.S.; Pollak, M.N.; Valsta, L.M.; Coronado, G.D.; Schwarz, Y.; Breymeyer, K.L.; Wang, C.; Wang, C.Y.; Lampe, J.W.; Neuhouser, M.L. Glycemic load effect on fasting and post-prandial serum glucose, insulin, IGF-1 and IGFBP-3 in a randomized, controlled feeding study. Eur. J. Clin. Nutr. 2012, 66, 1146–1152. [Google Scholar] [CrossRef]
- Runchey, S.S.; Valsta, L.M.; Schwarz, Y.; Wang, C.; Song, X.; Lampe, J.W.; Neuhouser, M.L. Effect of low- and high-glycemic load on circulating incretins in a randomized clinical trial. Metabolism 2013, 62, 188–195. [Google Scholar] [CrossRef]
- Barton, S.; Navarro, S.L.; Buas, M.F.; Schwarz, Y.; Gu, H.; Djukovic, D.; Raftery, D.; Kratz, M.; Neuhouser, M.L.; Lampe, J.W. Targeted plasma metabolome response to variations in dietary glycemic load in a randomized, controlled, crossover feeding trial in healthy adults. Food Funct. 2015, 6, 2949–2956. [Google Scholar] [CrossRef]
- Fu, B.C.; Randolph, T.W.; Lim, U.; Monroe, K.R.; Cheng, I.; Wilkens, L.R.; Le Marchand, L.; Hullar, M.A.; Lampe, J.W. Characterization of the gut microbiome in epidemiologic studies: The multiethnic cohort experience. Ann. Epidemiol. 2016, 26, 373–379. [Google Scholar] [CrossRef]
- Fu, B.C.; Randolph, T.W.; Lim, U.; Monroe, K.R.; Cheng, I.; Wilkens, L.R.; Le Marchand, L.; Lampe, J.W.; Hullar, M.A.J. Temporal variability and stability of the fecal microbiome: The Multiethnic Cohort Study. Cancer Epidemiol. Biomark. Prev. 2019, 28, 154–162. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar] [CrossRef]
- Integrative HMPRNC. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef]
- Keller, A.; Eng, J.; Zhang, N.; Li, X.J.; Aebersold, R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005, 1, 2005.0017. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Slagel, J.; Sun, Z.; Moritz, R.L. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteom. Clin. Appl. 2015, 9, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Gurdeep Singh, R.; Tanca, A.; Palomba, A.; Van der Jeugt, F.; Verschaffelt, P.; Uzzau, S.; Martens, L.; Dawyndt, P.; Mesuere, B. Unipept 4.0: Functional Analysis of Metaproteome Data. J. Proteome Res. 2019, 18, 606–615. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Dunaif, G.; Schneeman, B.O. The effect of dietary fiber on human pancreatic enzyme activity in vitro. Am. J. Clin. Nutr. 1981, 34, 1034–1035. [Google Scholar] [CrossRef]
- Walkowiak, J.; Wadolowska, L.; Szaflarska-Poplawska, A.; Lisowska, A.; Bugajewska, A.; Przyslawski, J. The elimination of meat from the diet selectively decreases pancreatic elastase secretion. Br. J. Nutr. 2007, 98, 154–158. [Google Scholar] [CrossRef][Green Version]
- Walkowiak, J.; Madry, E.; Lisowska, A.; Szaflarska-Poplawska, A.; Grzymislawski, M.; Stankowiak-Kulpa, H.; Przyslawski, J. Adaptive changes of pancreatic protease secretion to a short-term vegan diet: Influence of reduced intake and modification of protein. Br. J. Nutr. 2012, 107, 272–276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lovegrove, A.; Edwards, C.H.; De Noni, I.; Patel, H.; El, S.N.; Grassby, T.; Zielke, C.; Ulmius, M.; Nilsson, L.; Butterworth, P.J.; et al. Role of polysaccharides in food, digestion, and health. Crit. Rev. Food Sci. Nutr. 2017, 57, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, G.; Lundquist, I.; Ihse, I. Effect of dietary fiber on pancreatic enzyme activity in vitro. Gastroenterology 1982, 82 Pt 1, 918–924. [Google Scholar] [CrossRef]
- Dutta, S.K.; Hlasko, J. Dietary fiber in pancreatic disease: Effect of high fiber diet on fat malabsorption in pancreatic insufficiency and in vitro study of the interaction of dietary fiber with pancreatic enzymes. Am. J. Clin. Nutr. 1985, 41, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Hooda, S.; Boler, B.M.; Serao, M.C.; Brulc, J.M.; Staeger, M.A.; Boileau, T.W.; Dowd, S.E.; Fahey, G.C., Jr.; Swanson, K.S. 454 pyrosequencing reveals a shift in fecal microbiota of healthy adult men consuming polydextrose or soluble corn fiber. J. Nutr. 2012, 142, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.B.; Bruce, S.J.; Blondel-Lubrano, A.; Oguey-Araymon, S.; Beaumont, M.; Bourgeois, A.; Nielsen-Moennoz, C.; Vigo, M.; Fay, L.B.; Kochhar, S.; et al. A whole-grain cereal-rich diet increases plasma betaine, and tends to decrease total and LDL-cholesterol compared with a refined-grain diet in healthy subjects. Br. J. Nutr. 2011, 105, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Costabile, A.; Klinder, A.; Fava, F.; Napolitano, A.; Fogliano, V.; Leonard, C.; Gibson, G.R.; Tuohy, K.M. Whole-grain wheat breakfast cereal has a prebiotic effect on the human gut microbiota: A double-blind, placebo-controlled, crossover study. Br. J. Nutr. 2008, 99, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Finley, J.W.; Burrell, J.B.; Reeves, P.G. Pinto bean consumption changes SCFA profiles in fecal fermentations, bacterial populations of the lower bowel, and lipid profiles in blood of humans. J. Nutr. 2007, 137, 2391–2398. [Google Scholar] [CrossRef]
- Smith, S.C.; Choy, R.; Johnson, S.K.; Hall, R.S.; Wildeboer-Veloo, A.C.; Welling, G.W. Lupin kernel fiber consumption modifies fecal microbiota in healthy men as determined by rRNA gene fluorescent in situ hybridization. Eur. J. Nutr. 2006, 45, 335–341. [Google Scholar] [CrossRef]
- Johnson, S.K.; Chua, V.; Hall, R.S.; Baxter, A.L. Lupin kernel fibre foods improve bowel function and beneficially modify some putative faecal risk factors for colon cancer in men. Br. J. Nutr. 2006, 95, 372–378. [Google Scholar] [CrossRef]
- Tuohy, K.M.; Kolida, S.; Lustenberger, A.M.; Gibson, G.R. The prebiotic effects of biscuits containing partially hydrolysed guar gum and fructo-oligosaccharides—A human volunteer study. Br. J. Nutr. 2001, 86, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Hylla, S.; Gostner, A.; Dusel, G.; Anger, H.; Bartram, H.P.; Christl, S.U.; Kasper, H.; Scheppach, W. Effects of resistant starch on the colon in healthy volunteers: Possible implications for cancer prevention. Am. J. Clin. Nutr. 1998, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Lozupone, C.; Faust, K.; Raes, J.; Faith, J.J.; Frank, D.N.; Zaneveld, J.; Gordon, J.I.; Knight, R. Identifying genomic and metabolic features that can underline early successional and opportunistic lifestyles of human gut symbionts. Genome Res. 2012, 22, 1974–1984. [Google Scholar] [CrossRef]
- Bolca, S.; Van de Wiele, T.; Possemiers, S. Gut metabotypes govern health effects of dietary polyphenols. Curr. Opin. Biotechnol. 2013, 24, 220–225. [Google Scholar] [CrossRef]
- Heinzmann, S.S.; Merrifield, C.A.; Rezzi, S.; Kochhar, S.; Lindon, J.C.; Holmes, E.; Nicholson, J.K. Stability and robustness of human metabolic phenotypes in response to sequential food challenges. J. Proteome Res. 2012, 11, 643–655. [Google Scholar] [CrossRef]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Ndeh, D.; Rogowski, A.; Cartmell, A.; Luis, A.S.; Basle, A.; Gray, J.; Venditto, I.; Briggs, J.; Zhang, X.; Labourel, A.; et al. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature 2017, 544, 65–70. [Google Scholar] [CrossRef]
- Smiley, J.D.; Ashwell, G. Uronic acid metabolism in bacteria. III. Purification and properties of D-altronic acid and D-mannonic acid dehydrases in Escherichia coli. J. Biol Chem. 1960, 235, 1571–1575. [Google Scholar] [PubMed]
- Hickman, J.; Ashwell, G. Uronic acid metabolism in bacteria. II. Purification and properties of D-altronic acid and D-mannonic acid dehydrogenases in Escherichia coli. J. Biol Chem. 1960, 235, 1566–1570. [Google Scholar] [PubMed]
- Bach Knudsen, K.E. Microbial degradation of whole-grain complex carbohydrates and impact on short-chain fatty acids and health. Adv. Nutr. 2015, 6, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Di, Y.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Hague, A.; Elder, D.J.E.; Hicks, D.J.; Paraskeva, C. Apoptosis in colorectal tumor-cells—Induction by the short-chain fatty-acids butyrate, propionate and acetate and by the bile-salt deoxycholate. Int. J. Cancer 1995, 60, 400–406. [Google Scholar] [CrossRef]
- Jan, G.; Belzacq, A.S.; Haouzi, D.; Rouault, A.; Metivier, D.; Kroemer, G.; Brenner, C. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002, 9, 179–188. [Google Scholar] [CrossRef]
- Scheppach, W.; Bartram, H.P.; Richter, F. Role of short-chain fatty-acids in the prevention of colorectal-cancer. Eur. J. Cancer 1995, 31A, 1077–1080. [Google Scholar] [CrossRef]
- Campbell, J.M.; Fahey, G.C., Jr.; Wolf, B.W. Selected indigestible oligosaccharides affect large bowel mass, cecal and fecal short-chain fatty acids, pH and microflora in rats. J. Nutr. 1997, 127, 130–136. [Google Scholar] [CrossRef]
- Smiricky-Tjardes, M.R.; Grieshop, C.M.; Flickinger, E.A.; Bauer, L.L.; Fahey, G.C., Jr. Dietary galactooligosaccharides affect ileal and total-tract nutrient digestibility, ileal and fecal bacterial concentrations, and ileal fermentative characteristics of growing pigs. J. Anim. Sci. 2003, 81, 2535–2545. [Google Scholar] [CrossRef]
- De Angelis, M.; Montemurno, E.; Vannini, L.; Cosola, C.; Cavallo, N.; Gozzi, G.; Maranzano, V.; Di Cagno, R.; Gobbetti, M.; Gesualdo, L. Effect of Whole-Grain Barley on the Human Fecal Microbiota and Metabolome. Appl. Environ. Microbiol. 2015, 81, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; McCrae, S.I.; Charrier, C.; Flint, H.J. Organization of butyrate synthetic genes in human colonic bacteria: Phylogenetic conservation and horizontal gene transfer. FEMS Microbiol. Lett. 2007, 269, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J. Appl. Microbiol. 2007, 102, 1197–1208. [Google Scholar] [CrossRef]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef]
- Gibson, G.R.; Macfarlane, G.T.; Cummings, J.H. Sulphate reducing bacteria and hydrogen metabolism in the human large intestine. Gut 1993, 34, 437–439. [Google Scholar] [CrossRef]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Hansson, G.C. Role of mucus layers in gut infection and inflammation. Curr. Opin. Microbiol. 2012, 15, 57–62. [Google Scholar] [CrossRef]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. mBio 2014, 5, e00889. [Google Scholar] [CrossRef]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microb. 2000, 66, 1654–1661. [Google Scholar] [CrossRef]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Matthies, C.; Schink, B. Fermentative degradation of glutarate via decarboxylation by newly isolated strictly anaerobic-bacteria. Arch. Microbiol. 1992, 157, 290–296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matthies, C.; Schink, B. Reciprocal isomerization of butyrate and isobutyrate by the strictly anaerobic bacterium strain-wog13 and methanogenic isobutyrate degradation by a defined triculture. Appl. Environ. Microbiol. 1992, 58, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Roeder, J.; Schink, B. Syntrophic degradation of Cadaverine by a defined methanogenic coculture. Appl. Environ. Microbiol. 2009, 75, 4821–4828. [Google Scholar] [CrossRef] [PubMed]
- Gharbia, S.E.; Shah, H.N. Pathways of glutamate catabolism among Fusobacterium Species. J. Gen. Microbiol. 1991, 137, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, A.; Cinkaya, I.; Linder, D.; Huisman, G.; Buckel, W. Fermentation of 4-aminobutyrate by Clostridium aminobutyricum: Cloning of two genes involved in the formation and dehydration of 4-hydroxybutyryl-CoA. Arch. Microbiol. 2000, 174, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W. Unusual enzymes involved in five pathways of glutamate fermentation. Appl. Microbiol Biotechnol. 2001, 57, 263–273. [Google Scholar] [CrossRef]
- Kreimeyer, A.; Perret, A.; Lechaplais, C.; Vallenet, D.; Medigue, C.; Salanoubat, M.; Weissenbach, J. Identification of the last unknown genes in the fermentation pathway of lysine. J. Biol. Chem. 2007, 282, 7191–7197. [Google Scholar] [CrossRef]
- Potrykus, J.; White, R.L.; Bearne, S.L. Proteomic investigation of amino acid catabolism in the indigenous gut anaerobe Fusobacterium varium. Proteomics 2008, 8, 2691–2703. [Google Scholar] [CrossRef]
- Uematsu, H.; Hoshino, E. Degradation of arginine and other amino acids by Eubacterium nodatum ATCC 33099. Microb. Ecol. Health Dis. 1996, 9, 305–311. [Google Scholar] [CrossRef][Green Version]
- Hippe, B.; Zwielehner, J.; Liszt, K.; Lassl, C.; Unger, F.; Haslberger, A.G. Quantification of butyryl CoA:acetate CoA-transferase genes reveals different butyrate production capacity in individuals according to diet and age. FEMS Microbiol. Lett. 2011, 316, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA: Acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Gao, J.; Rizzo, M.; Harrison, T.; Tiedje, J.M. Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J. 2015, 9, 832–843. [Google Scholar] [CrossRef]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Kado, Y.; Takada, T.; Matsumoto, K.; Tanaka, R. Quantitative PCR with 16S rRNA-gene-targeted species-specific primers for analysis of human intestinal bifidobacteria. Appl. Environ. Microbiol. 2004, 70, 167–173. [Google Scholar] [CrossRef]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Takada, T.; Tanaka, R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol. 2004, 70, 7220–7228. [Google Scholar] [CrossRef]
- Wang, R.F.; Cao, W.W.; Cerniglia, C.E. PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Appl. Environ. Microbiol. 1996, 62, 1242–1247. [Google Scholar] [CrossRef]
- Hosseini, E.; Grootaert, C.; Verstraete, W.; Van de Wiele, T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr. Rev. 2011, 69, 245–258. [Google Scholar] [CrossRef]
- Cheng, H.H.; Lai, M.H. Fermentation of resistant rice starch produces propionate reducing serum and hepatic cholesterol in rats. J. Nutr. 2000, 130, 1991–1995. [Google Scholar] [CrossRef]
- Ximenes, H.M.; Hirata, A.E.; Rocha, M.S.; Curi, R.; Carpinelli, A.R. Propionate inhibits glucose-induced insulin secretion in isolated rat pancreatic islets. Cell Biochem. Funct. 2007, 25, 173–178. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.Z.; et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 2014, 147, 1055–1063.e8. [Google Scholar] [CrossRef] [PubMed]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Imlay, J. Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch. Biochem. Biophys. 2012, 525, 145–160. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Linden, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin glycan foraging in the human gut microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Liu, X.W.; Liu, J.P.; Yang, X.Y.; Lu, F.G. Ulcerative colitis as a polymicrobial infection characterized by sustained broken mucus barrier. World J. Gastroenterol. 2014, 20, 9468–9475. [Google Scholar] [CrossRef]
- Sheng, Y.H.; Hasnain, S.Z.; Florin, T.H.; McGuckin, M.A. Mucins in inflammatory bowel diseases and colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 28–38. [Google Scholar] [CrossRef]
- Kolmeder, C.A.; de Vos, W.M. Metaproteomics of our microbiome—Developing insight in function and activity in man and model systems. J. Proteom. 2014, 97, 3–16. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.M.; Chu, K.; Szeto, E.; Palaniappan, K.; Jacob, B.; Ratner, A.; Liolios, K.; Pagani, I.; Huntemann, M.; et al. IMG/M-HMP: A metagenome comparative analysis system for the Human Microbiome Project. PLoS ONE 2012, 7, e40151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PEPTIDE COUNTS Average (SEM) | ||||
|---|---|---|---|---|
| EC number | Enzyme Name | RG | WG | p value* |
| Higher after WG Diet | ||||
| 1.1.1.- | Oxidoreductases Acting on the CH-OH group of donors with NAD(+) or NADP(+) as acceptor | 17.2 (4) | 25.2 (5.4) | 0.002584 |
| 1.1.1.1 | Alcohol dehydrogenase | 25.9 (11.8) | 35.1 (9.6) | 0.000514 |
| 1.1.1.157 | 3-hydroxybutyryl-CoA dehydrogenase | 23.1 (12.8) | 37.9 (13.7) | <0.000001 |
| 1.1.1.35 | 3-hydroxyacyl-CoA dehydrogenase | 18.1 (12.4) | 33.3 (11.7) | <0.000001 |
| 1.1.1.37 | Malate dehydrogenase | 21.4 (14.2) | 39 (11.9) | <0.000001 |
| 1.1.1.58 | Tagaturonate reductase | 12.7 (8.3) | 26.3 (9.5) | <0.000001 |
| 1.1.1.69 | Gluconate 5-dehydrogenase | 14.2 (7.3) | 23.9 (6.8) | 0.000272 |
| 1.1.1.86 | Ketol-acid reductoisomerase (NADP(+)) | 52.3 (19.3) | 60.8 (13.4) | 0.001469 |
| 1.17.7.4 | 4-hydroxy-3-methylbut-2-enyl diphosphate reductase | 21.7 (10.6) | 30.8 (7.2) | 0.0006 |
| 1.2.1.- | Oxidoreductases Acting on the aldehyde or oxo group of donors with NAD(+) or NADP(+) as acceptor | 189.1 (51.9) | 214.2 (44.2) | <0.000001 |
| 1.2.7.- | Oxidoreductases Acting on the aldehyde or oxo group of donors with an iron-sulfur protein as acceptor | 159.2 (64.8) | 185.8 (37.2) | <0.000001 |
| 1.3.8.1 | Butyryl-CoA dehydrogenase | 47.7 (16.4) | 61.9 (19) | <0.000001 |
| 2.2.1.1 | Transketolase | 22.3 (13.1) | 36.4 (13.1) | <0.000001 |
| 2.2.1.2 | Transaldolase | 20.8 (7.1) | 29.2 (13.7) | 0.001469 |
| 2.3.1.54 | Formate C-acetyltransferase | 36.1 (23.3) | 44 (24.1) | 0.002964 |
| 2.3.1.8 | Phosphate acetyltransferase | 17.3 (11.4) | 28.3 (8.8) | 0.000034 |
| 2.3.1.9 | Acetyl-CoA C-acetyltransferase | 48.7 (17.2) | 58.4 (10.5) | 0.000231 |
| 2.4.1.1 | Glycogen phosphorylase | 37.6 (13.6) | 48.4 (14.6) | 0.000041 |
| 2.5.1.47 | Cysteine synthase | 2.1 (3.1) | 11.9 (7.9) | 0.000231 |
| 2.5.1.49 | O-acetylhomoserine aminocarboxypropyltransferase | 5.3 (5) | 14.2 (6.2) | 0.000814 |
| 2.6.1.52 | Phosphoserine transaminase | 22.2 (5.9) | 30.3 (7.9) | 0.00225 |
| 2.7.1.92 | 5-dehydro-2-deoxygluconokinase | 16.8 (9.1) | 24.6 (7.1) | 0.003394 |
| 2.7.2.1 | Acetate kinase | 27.8 (9.2) | 37.4 (9.6) | 0.000272 |
| 2.7.2.3 | Phosphoglycerate kinase | 75.3 (30.6) | 107.3 (23.7) | <0.000001 |
| 2.7.7.27 | Glucose-1-phosphate adenylyltransferase | 22.9 (15.6) | 31.1 (10.2) | 0.001955 |
| 2.7.7.6 | DNA-directed RNA polymerase | 124.1 (30.9) | 136.3 (23.6) | 0.000004 |
| 2.7.7.8 | Polyribonucleotide nucleotidyltransferase | 28.8 (14.1) | 36.8 (11.6) | 0.002584 |
| 2.7.9.1 | Pyruvate, phosphate dikinase | 130.9 (35.2) | 168.9 (40.3) | <0.000001 |
| 3.5.-.- | Hydrolases Acting on carbon-nitrogen bonds, other than peptide bonds | 6.6 (6.2) | 15.1 (11.3) | 0.001271 |
| 3.6.3.14 | H(+)-transporting two-sector ATPase | 33.8 (13.4) | 47.1 (16.4) | <0.000001 |
| 3.6.5.3 | Protein-synthesizing GTPase | 93.8 (25.9) | 112.7 (30.6) | <0.000001 |
| 4.1.1.49 | Phosphoenolpyruvate carboxykinase (ATP) | 126.1 (50.6) | 157 (26.1) | <0.000001 |
| 4.1.2.13 | Fructose-bisphosphate aldolase | 65.2 (25.3) | 92.8 (20.9) | <0.000001 |
| 4.2.1.11 | Phosphopyruvate hydratase | 24.7 (13.6) | 32.9 (14.8) | 0.001955 |
| 4.2.1.17 | Enoyl-CoA hydratase (altronate dehydratase) | 15.2 (7.4) | 27.4 (7.8) | 0.000004 |
| 4.2.1.55 | 3-hydroxybutyryl-CoA dehydratase | 13.7 (7.2) | 23.8 (6.9) | 0.00014 |
| 4.4.1.11 | Methionine gamma-lyase | 5.4 (4.1) | 14.1 (6.4) | 0.001097 |
| 5.3.1.1 | Triose-phosphate isomerase | 43.2 (15) | 56.7 (12.2) | <0.000001 |
| 5.3.1.9 | Glucose-6-phosphate isomerase | 29.1 (11.8) | 42.1 (11.6) | <0.000001 |
| 5.4.2.2 | Phosphoglucomutase (alpha-d-glucose-1,6-bisphosphate-dependent) | 33.4 (15.7) | 50.7 (19.3) | <0.000001 |
| 5.4.2.8 | Phosphomannomutase | 13.7 (8.1) | 22.4 (9.6) | 0.000946 |
| 5.4.99.2 | Methylmalonyl-CoA mutase | 20.8 (15.4) | 28.6 (15.5) | 0.003394 |
| 6.3.1.2 | Glutamine synthetase | 24.9 (18.7) | 37.3 (20.5) | 0.000003 |
| 6.3.2.6 | Phosphoribosylaminoimidazolesuccinocarboxamide synthase purine | 20.8 (7) | 29.4 (11.2) | 0.001097 |
| 6.3.5.5 | Carbamoyl-phosphate synthase (glutamine-hydrolyzing) | 22.2 (9.5) | 36.4 (7.2) | <0.000001 |
| Higher after RG Diet | ||||
| 1.11.1.1 | NADH peroxidase | 72.8 (29.4) | 57.8 (20.1) | <0.000001 |
| 3.2.1.23 | Beta-galactosidase | 46.3 (21.1) | 37.9 (14.4) | 0.001469 |
| 3.2.1.97 | Endo-alpha-N-acetylgalactosaminidase | 21.2 (20.8) | 7.4 (6.2) | <0.000001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, S.; Hullar, M.A.J.; Lai, L.A.; Peng, H.; May, D.H.; Noble, W.S.; Raftery, D.; Navarro, S.L.; Neuhouser, M.L.; Lampe, P.D.; et al. Gut Microbial Protein Expression in Response to Dietary Patterns in a Controlled Feeding Study: A Metaproteomic Approach. Microorganisms 2020, 8, 379. https://doi.org/10.3390/microorganisms8030379
Pan S, Hullar MAJ, Lai LA, Peng H, May DH, Noble WS, Raftery D, Navarro SL, Neuhouser ML, Lampe PD, et al. Gut Microbial Protein Expression in Response to Dietary Patterns in a Controlled Feeding Study: A Metaproteomic Approach. Microorganisms. 2020; 8(3):379. https://doi.org/10.3390/microorganisms8030379
Chicago/Turabian StylePan, Sheng, Meredith A. J. Hullar, Lisa A. Lai, Hong Peng, Damon H. May, William S. Noble, Daniel Raftery, Sandi L. Navarro, Marian L. Neuhouser, Paul D. Lampe, and et al. 2020. "Gut Microbial Protein Expression in Response to Dietary Patterns in a Controlled Feeding Study: A Metaproteomic Approach" Microorganisms 8, no. 3: 379. https://doi.org/10.3390/microorganisms8030379
APA StylePan, S., Hullar, M. A. J., Lai, L. A., Peng, H., May, D. H., Noble, W. S., Raftery, D., Navarro, S. L., Neuhouser, M. L., Lampe, P. D., Lampe, J. W., & Chen, R. (2020). Gut Microbial Protein Expression in Response to Dietary Patterns in a Controlled Feeding Study: A Metaproteomic Approach. Microorganisms, 8(3), 379. https://doi.org/10.3390/microorganisms8030379