There is No Distinctive Gut Microbiota Signature in the Metabolic Syndrome: Contribution of Cardiovascular Disease Risk Factors and Associated Medication

 ,
,  , and
, and 
Abstract
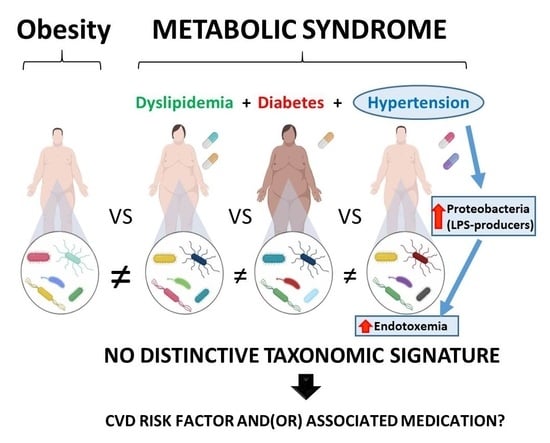
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Sampling Procedure
2.3. Determination of Serobiochemical Variables and Lipopolysaccharide-Binding Protein (LBP)
2.4. Gut Microbiota Analysis
2.5. Short-Chain Fatty Acids (SCFAs) Determination
2.6. Statistical Analysis
3. Results
3.1. Demographic Characteristics and Laboratory Values of the Population Study
3.2. Alpha-diversity Indexes of the Gut Microbiota
3.3. Comparison of Taxonomic Signatures in Obese Subjects and MetS Patients
3.4. Firmicutes to Bacteroidetes ratio (F/B) in OB and MetS Patients
3.5. Metabolic Function Prediction in OB and MetS Microbiota Profiles
3.6. Taxonomic Signatures in MetS Patients Grouped by Pharmacological Treatments
3.7. Hypertension Correlates with Higher Plasma LBP Values in MetS
3.8. Lower SCFA Production Correlates with Lipid-lowering (LL) Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fei, N.; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013, 7, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Lin, C.S.; Lu, C.C.; Martel, J.; Ko, Y.F.; Ojcius, D.M.; Tseng, S.F.; Wu, T.R.; Chen, Y.Y.; Young, J.D.; et al. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 2015, 6, 7489. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry. 2016, 21, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Borgen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Le Roy, T.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Haro, C.; García-Carpintero, S.; Rangel-Zúñiga, O.A.; Alcalá-Díaz, J.F.; Landa, B.B.; Clemente, J.C.; Pérez-Martínez, P.; López-Miranda, J.; Pérez-Jiménez, F.; Camargo, A. Consumption of two healthy dietary patterns restored microbiota dysbiosis in obese patients with metabolic dysfunction. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Cani, P.D. Microbiota and metabolites in metabolic diseases. Nat. Rev. Endocrinol. 2019, 15, 69–70. [Google Scholar] [CrossRef]
- Zhang, Z.; Mocanu, V.; Cai, C.; Dang, J.; Slater, L.; Deehan, E.C.; Walter, J.; Madsen, K.L. Impact of fecal microbiota transplantation on obesity and metabolic syndrome-A systematic review. Nutrients 2019, 11, 2291. [Google Scholar] [CrossRef] [PubMed]
- Hooks, K.B.; O’Malley, M.A. Dysbiosis and Its Discontents. mbio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J.; Alexander, J.L.; Merrifield, C.A.; Cunningham, D.; Jobin, C.; Brown, R.; Alverdy, J.; O’Keefe, S.J.; Gaskins, H.R.; Teare, J.; et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut 2019, 68, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Lee, C.J.; Sears, C.L.; Maruthur, N. Gut microbiome and its role in obesity and insulin resistance. Ann. N. Y. Acad. Sci. 2020, 1461, 37–52. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Finucane, M.M.; Sharpton, T.J.; Laurent, T.J.; Pollard, K.S. A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- He, Y.; Wu, W.; Zheng, H.M.; Li, P.; McDonald, D.; Sheng, H.F.; Chen, M.X.; Chen, Z.H.; Ji, G.Y.; Zheng, Z.D.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Graham, C.; Mullen, A.; Whelan, K. Obesity and the gastrointestinal microbiota: A review of associations and mechanisms. Nutr. Rev. 2015, 73, 376–385. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Bremer, A.A.; Lustig, R.H. What is metabolic syndrome, and why are children getting it? Ann. N. Y. Acad. Sci. 2013, 1281, 123–140. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—a new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Zupancic, M.L.; Cantarel, B.L.; Liu, Z.; Drabek, E.F.; Ryan, K.A.; Cirimotich, S.; Jones, C.; Knight, R.; Walters, W.A.; Knights, D.; et al. Analysis of the gut microbiota in the old order Amish and its relation to the metabolic syndrome. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Santos-Marcos, J.A.; Pérez-Jiménez, F.; Camargo, A. The role of diet and intestinal microbiota in the development of metabolic syndrome. J. Nutr. Biochem. 2019, 70, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Invest. 2019, 129, 4050–4057. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Sánchez-Alcoholado, L.; Pérez-Martínez, P.; Andrés-Lacueva, C.; Cardona, F.; Tinahones, F.; Queipo-Ortuño, M.I. Red wine polyphenols modulate fecal microbiota and reduce markers of the metabolic syndrome in obese patients. Food Funct. 2016, 7, 1775–1787. [Google Scholar] [CrossRef]
- Haro, C.; García-Carpintero, S.; Alcalá-Díaz, J.F.; Gómez-Delgado, F.; Delgado-Lista, J.; Pérez-Martínez, P.; Rangel-Zúñiga, O.A.; Quintana-Navarro, G.M.; Landa, B.B.; Clemente, J.C.; et al. The gut microbial community in metabolic syndrome patients is modified by diet. J. Nutr. Biochem. 2016, 27, 27–31. [Google Scholar] [CrossRef]
- Festi, D.; Schiumerini, R.; Eusebi, L.H.; Marasco, G.; Taddia, M.; Colecchia, A. Gut microbiota and metabolic syndrome. World J. Gastroenterol. 2014, 20, 16079–16094. [Google Scholar] [CrossRef]
- Wong, S.K.; Chin, K.Y.; Suhaimi, F.H.; Fairus, A.; Ima-Nirwana, S. Animal models of metabolic syndrome: A review. Nutr. Metab. (Lond.) 2016, 13, 65. [Google Scholar] [CrossRef]
- Bouter, K.E.; van Raalte, D.H.; Groen, A.K.; Nieuwdorp, M. Role of the gut microbiome in the pathogenesis of obesity and obesity-related metabolic dysfunction. Gastroenterology 2017, 152, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Romo-Vaquero, M.; Cortés-Martín, A.; Loria-Kohen, V.; Ramírez-de-Molina, A.; García-Mantrana, I.; Collado, M.C.; Espín, J.C.; Selma, M.V. Deciphering the human gut microbiome of urolithin metabotypes: association with enterotypes and potential cardiometabolic health implications. Mol. Nutr. Food Res. 2019, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, T.J. Limitations of the Limulus amebocyte lysate test in demonstrating circulating lipopolysaccharides. Ann. N. Y. Acad. Sci. 1998, 851, 416–421. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Romo-Vaquero, M.; García-Villalba, R.; Cortés-Martín, A.; Selma, M.V.; Espín, J.C. The endotoxemia marker lipopolysaccharide-binding protein is reduced in overweight-obese subjects consuming pomegranate extract by modulating the gut microbiota: A randomized clinical trial. Mol. Nutr. Food Res. 2018, 62, 1800160. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega-Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- García-Villalba, R.; Giménez-Bastida, J.A.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Espín, J.C.; Larrosa, M. Alternative method for gas chromatography-mass spectrometry analysis of short-chain fatty acids in faecal samples. J. Sep. Sci. 2012, 35, 1906–1913. [Google Scholar] [CrossRef]
- Pérez-Martínez, P.; Mikhailidis, D.P.; Athyros, V.G.; Bullo, M.; Couture, P.; Covas, M.I.; de Koning, L.; Delgado-Lista, J.; Díaz-López, A.; Drevon, C.A.; et al. Lifestyle recommendations for the prevention and management of metabolic syndrome: An international panel recommendation. Nutr. Rev. 2017, 75, 307–326. [Google Scholar] [CrossRef]
- Zeng, Q.; Li, D.; He, Y.; Li, Y.; Yang, Z.; Zhao, X.; Liu, Y.; Wang, Y.; Sun, J.; Feng, X.; et al. Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 2019, 9, 13424. [Google Scholar] [CrossRef]
- Ahmad, A.; Yang, W.; Chen, G.; Shafiq, M.; Javed, S.; Ali Zaidi, S.S.; Shahid, R.; Liu, C.; Bokhari, H. Analysis of gut microbiota of obese individuals with type 2 diabetes and healthy individuals. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Tandon, D.; Haque, M.M.; Saravanan, R.; Shaikh, S.; Sriram, P.; Dubey, A.K.; Mande, S.S. A snapshot of gut microbiota of an adult urban population from Western region of India. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Lelouvier, B.; Servant, F.; Lluch, J.; Burcelin, R.; Bongard, V.; Elbaz, M. Blood microbiota modification after myocardial infarction depends upon low-density lipoprotein cholesterol levels. J. Am. Heart Assoc. 2019, 8, e011797. [Google Scholar] [CrossRef]
- Caparrós-Martín, J.A.; Lareu, R.R.; Ramsay, J.P.; Peplies, J.; Reen, F.J.; Headlam, H.A.; Ward, N.C.; Croft, K.D.; Newsholme, P.; Hughes, J.D.; et al. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome 2017, 5, 95. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.; An, J.; Song, Y.; Lee, C.K.; Kim, K.; Kong, H. Alterations in gut microbiota by statin therapy and possible intermediate effects on hyperglycemia and hyperlipidemia. Front. Microbiol. 2019, 10, 1947. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Bryrup, T.; Thomsen, C.W.; Kern, T.; Allin, K.H.; Brandslund, I.; Jørgensen, N.R.; Vestergaard, H.; Hansen, T.; Hansen, T.H.; Pedersen, O.; et al. Metformin-induced changes of the gut microbiota in healthy young men: Results of a non-blinded, one-armed intervention study. Diabetologia 2019, 62, 1024–1035. [Google Scholar] [CrossRef]
- Elbere, I.; Kalnina, I.; Silamikelis, I.; Konrade, I.; Zaharenko, L.; Sekace, K.; Radovica-Spalvina, I.; Fridmanis, D.; Gudra, D.; Pirags, V.; et al. Association of metformin administration with gut microbiome dysbiosis in healthy volunteers. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Sun, S.; Lulla, A.; Sioda, M.; Winglee, K.; Wu, M.C.; Jacobs, D.R. Jr.; Shikany, J.M.; Lloyd-Jones, D.M.; Launer, L.J.; Fodor, A.A.; et al. Gut microbiota composition and blood pressure. Hypertension 2019, 73, 998–1006. [Google Scholar] [CrossRef]
- Robles-Vera, I.; Toral, M.; de la Visitación, N.; Sánchez, M.; Gómez-Guzmán, M.; Muñoz, R.; Algieri, F.; Vezza, T.; Jiménez, R.; Gálvez, J.; et al. Losartan-induced gut microbial changes has an antihypertensive effect. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Pevsner-Fischer, M.; Blacher, E.; Tatirovsky, E.; Ben-Dov, I.Z.; Elinav, E. The gut microbiome and hypertension. Curr. Opin. Nephrol. Hypertens. 2017, 26, 1–8. [Google Scholar] [CrossRef]
- Wang, J.; Gu, X.; Yang, J.; Wei, Y.; Zhao, Y. Gut microbiota dysbiosis and increased plasma LPS and TMAO levels in patients with preeclampsia. Front. Cell Infect. Microbiol. 2019, 9, 409. [Google Scholar] [CrossRef]
- Chang, Y.; Chen, Y.; Zhou, Q.; Wang, C.; Chen, L.; Di, W.; Zhang, Y. Short-chain fatty acids accompanying changes in the gut microbiome contribute to the development of hypertension in patients with preeclampsia. Clin. Sci. (Lond.) 2020, 134, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes. 2014, 5, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Markó, L.; Höges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Jin, M.; Kalainy, S.; Baskota, N.; Chiang, D.; Deehan, E.C.; McDougall, C.; Tandon, P.; Martínez, I.; Cervera, C.; Walter, J.; et al. Faecal microbiota from patients with cirrhosis has a low capacity to ferment non-digestible carbohydrates into short-chain fatty acids. Liver Int. 2019, 39, 1437–1447. [Google Scholar] [CrossRef]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of mucosal immune tesponse, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [PubMed]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Women (n = 24) | Men (n = 26) | p | |
|---|---|---|---|
| Age | 55.0 (35.0−74.0) | 56.0 (29.0−76.0) | 0.736 |
| BMI (kg/m2) | 33.6 (30−48.6) | 32.9 (30.4−45.3) | 0.573 |
| Drug treatments: | |||
| AD-consumers | 20 (40%) | 22 (44%) | |
| LL-consumers | 19 (38%) | 20 (40%) | |
| HP-consumers | 17 (34%) | 19 (38%) | |
| Systolic BP (mm Hg) | 136.0 (96.0−190.0) | 134.0 (99.0−201.0) | 0.165 |
| Diastolic BP (mm Hg) | 79.0 (55.0−102.0) | 82.0 (60.0−96.0) | 0.817 |
| Tchol (mg/dL) | 186.0 (116.0−249.0) | 158.0 (106.0−269.0) | 0.077 |
| LDLc (mg/dL) | 105.3 (49.0−163.0) | 85.5 (46.0−163.0) | 0.079 |
| HDLc (mg/dL) | 51.0 (28.5−85.5) | 46.0 (30.0−65.0) | 0.023 * |
| Triglycerides (mg/dL) | 118.0 (48.0−479.0) | 149.0 (64.0−611.0) | 0.065 |
| Glucose (mg/dL) | 95.5 (79.0−194.0) | 114.0 (73.0−232.0) | 0.307 |
| Insulin (μUl/mL) | 9.7 (2.9−34.3) | 15.9 (6.4−74.8) | 0.027 * |
| HOMA-IR (U) | 2.7 (0.9−9.3) | 5.0 (1.4−15.1) | 0.012 * |
| Bilirubin (U/L) | 0.4 (0.2−0.7) | 0.5 (0.2−1.1) | 0.257 |
| Protein (U/L) | 7.0 (6.5−8.1) | 7.2 (6.4−8.5) | 0.682 |
| GGT (U/L) | 17.0 (8.0−147.0) | 31.0 (12.0−283.0) | 0.012 * |
| AST (U/L) | 18.0 (12.0−43.0) | 21.0 (11.0−64.0) | 0.125 |
| ALT (U/L) | 19.0 (11.0−59.0) | 23.0 (9.0−68.0) | 0.105 |
| ALP (U/L) | 74.0 (44.0−279.0) | 73.0 (38.0−317.0) | 0.957 |
| LDH (U/L) | 175.0 (122.0−354.0) | 179.0 (139.0−524.0) | 0.511 |
| Calcium (mg/dL) | 9.5 (8.9−10.8) | 9.6 (8.7−10.4) | 0.920 |
| Phosphorus (mg/dL) | 3.5 (2.3−4.6) | 3.3 (1.9−4.3) | 0.174 |
| Sodium (mEq/L) | 141.0 (137.0−147.0) | 141.0 (137.0−145.0) | 0.444 |
| Potassium (mEq/L) | 4.5 (4.3−5.4) | 4.5 (3.9−5.5) | 0.260 |
| Chlorine (mEq/L) | 101.5 (95.0−105.0) | 103.0 (99.0−105.0) | 0.211 |
| Albumin (g/dL) | 4.5 (4.0−5.0) | 4.6 (3.9−5.1) | 0.687 |
| Urea (mg/dL) | 39.0 (23.0−87.0) | 34.0 (22.0−88.0) | 0.624 |
| Creatinine (mg/dL) | 0.7 (0.5−2.0) | 0.9 (0.6−1.9) | 0.024 * |
| LBP (µg/mL) | 7.9 (7.2−9.2) | 8.1 (7.1−10.4) | 0.022 * |
| Acetic acid (µmol/L) | 1249.6 (414.0−3432.4) | 1697.1 (264.0−3373.2) | 0.219 |
| Propionic acid (µmol/L) | 960.2 (292.5−3176.9) | 1228.0 (167.9−2749.6) | 0.135 |
| Isobutyric acid (µmol/L) | 127.5 (13.6−410.0) | 167.9 (50.8−396.4) | 0.180 |
| Butyric acid (µmol/L) | 1100.9 (241.5−3218.2) | 1187.7 (148.1−3608.1) | 0.204 |
| Isovaleric acid (µmol/L) | 180.7 (14.1−760.9) | 264.1 (78.6−676.5) | 0.210 |
| Valeric acid (µmol/L) | 194.7 (20.0−415.1) | 284.2 (17.3−608.5) | 0.166 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Martín, A.; Iglesias-Aguirre, C.E.; Meoro, A.; Selma, M.V.; Espín, J.C. There is No Distinctive Gut Microbiota Signature in the Metabolic Syndrome: Contribution of Cardiovascular Disease Risk Factors and Associated Medication. Microorganisms 2020, 8, 416. https://doi.org/10.3390/microorganisms8030416
Cortés-Martín A, Iglesias-Aguirre CE, Meoro A, Selma MV, Espín JC. There is No Distinctive Gut Microbiota Signature in the Metabolic Syndrome: Contribution of Cardiovascular Disease Risk Factors and Associated Medication. Microorganisms. 2020; 8(3):416. https://doi.org/10.3390/microorganisms8030416
Chicago/Turabian StyleCortés-Martín, Adrián, Carlos E. Iglesias-Aguirre, Amparo Meoro, María Victoria Selma, and Juan Carlos Espín. 2020. "There is No Distinctive Gut Microbiota Signature in the Metabolic Syndrome: Contribution of Cardiovascular Disease Risk Factors and Associated Medication" Microorganisms 8, no. 3: 416. https://doi.org/10.3390/microorganisms8030416
APA StyleCortés-Martín, A., Iglesias-Aguirre, C. E., Meoro, A., Selma, M. V., & Espín, J. C. (2020). There is No Distinctive Gut Microbiota Signature in the Metabolic Syndrome: Contribution of Cardiovascular Disease Risk Factors and Associated Medication. Microorganisms, 8(3), 416. https://doi.org/10.3390/microorganisms8030416