Mechanisms Underlying the Rhizosphere-To-Rhizoplane Enrichment of Cellvibrio Unveiled by Genome-Centric Metagenomics and Metatranscriptomics

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Metagenome-Assembled Genome Extraction and Curation
2.2. Taxonomic Classification
2.3. Genome Annotation
2.4. Comparative Genomics Analysis of Cellvibrio sp. Bin79 with Related Cellvibrio Genomes
2.5. Differential Gene Expression Analysis of Bin79 Between Rhizosphere and Rhizoplane Niches
3. Results and Discussion
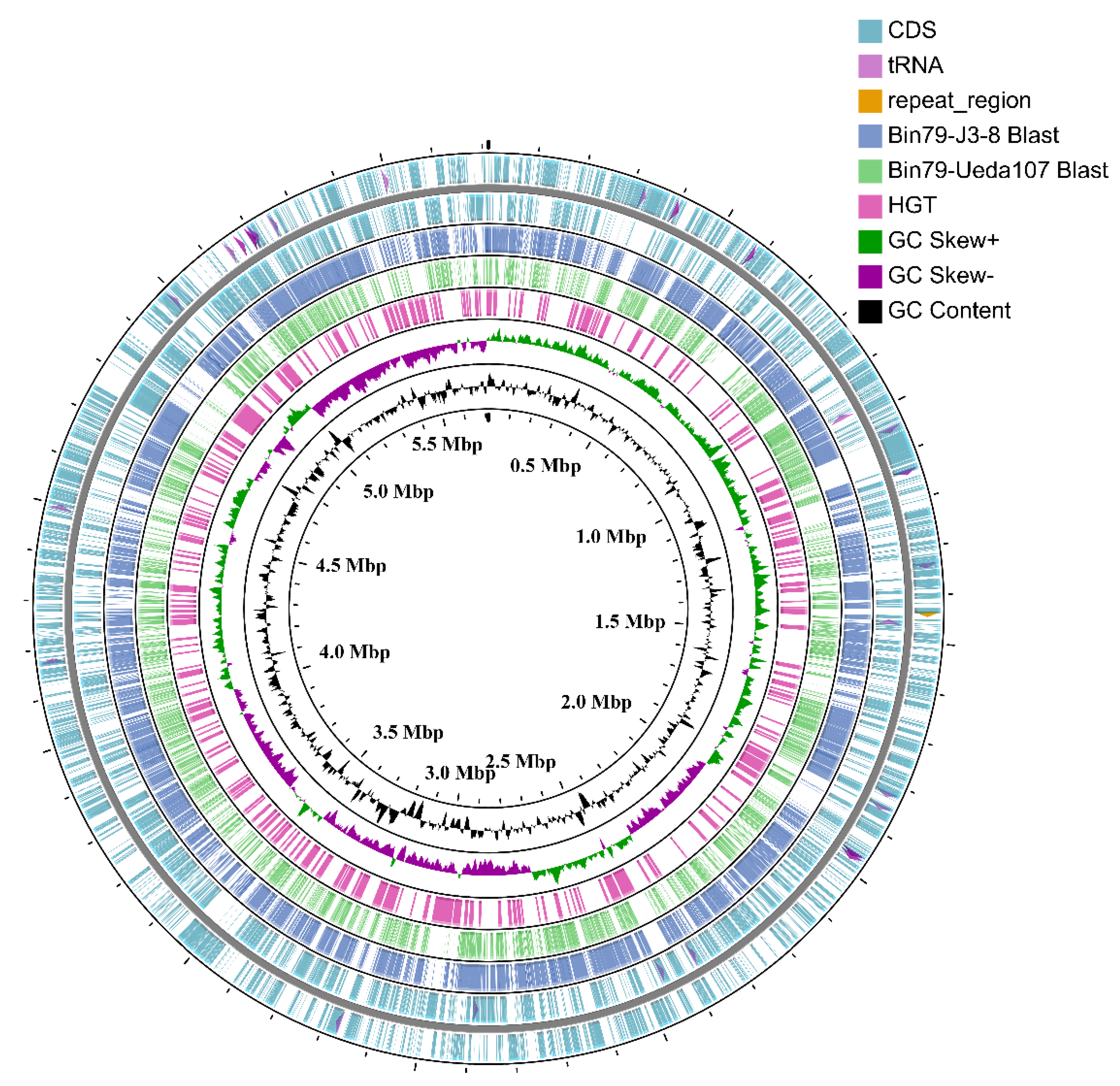
3.1. Recovery and Curation of the Almost Complete Metagenome-Assembled Genome Bin79
3.2. Phylogenetic Analysis of Bin79
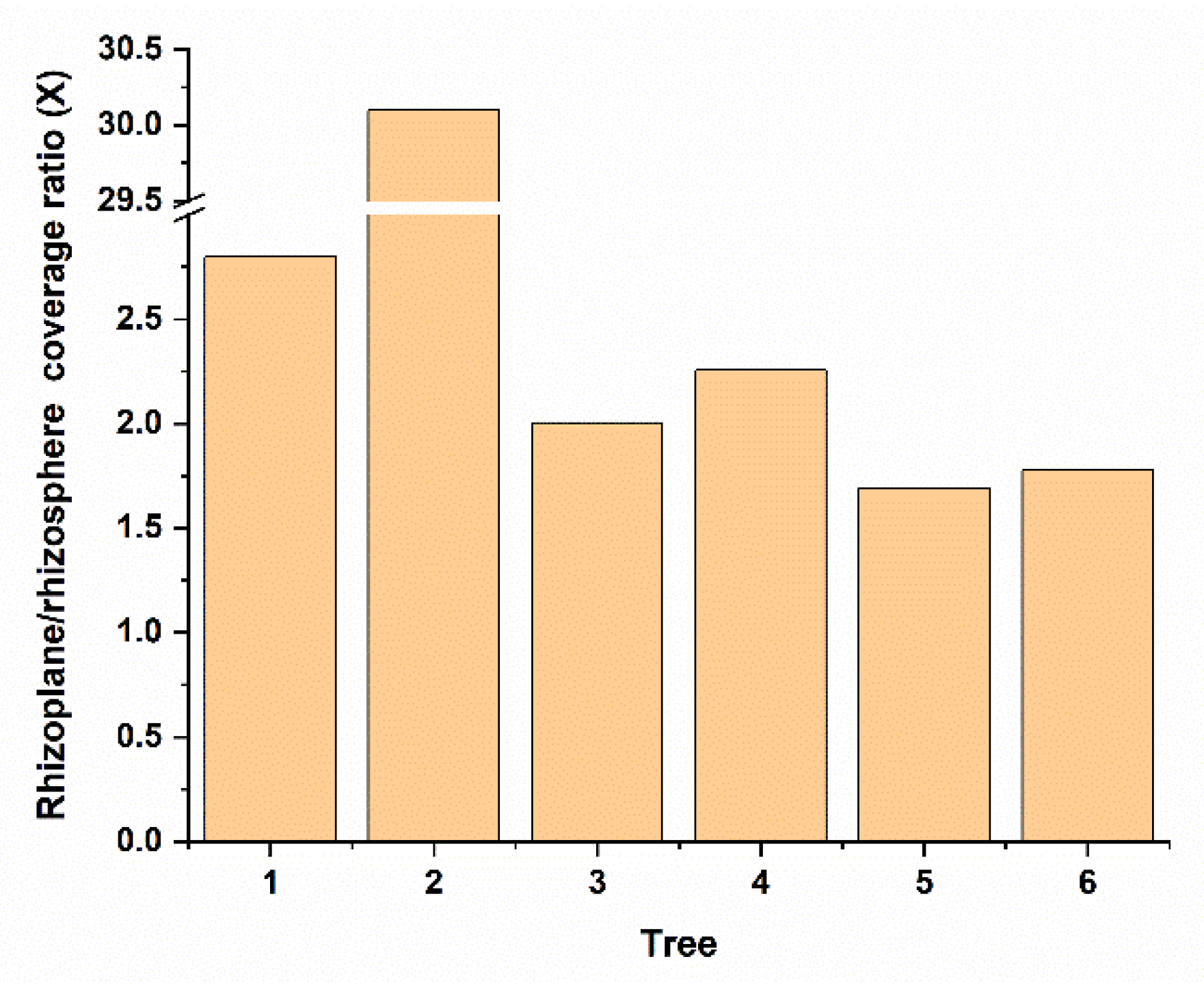
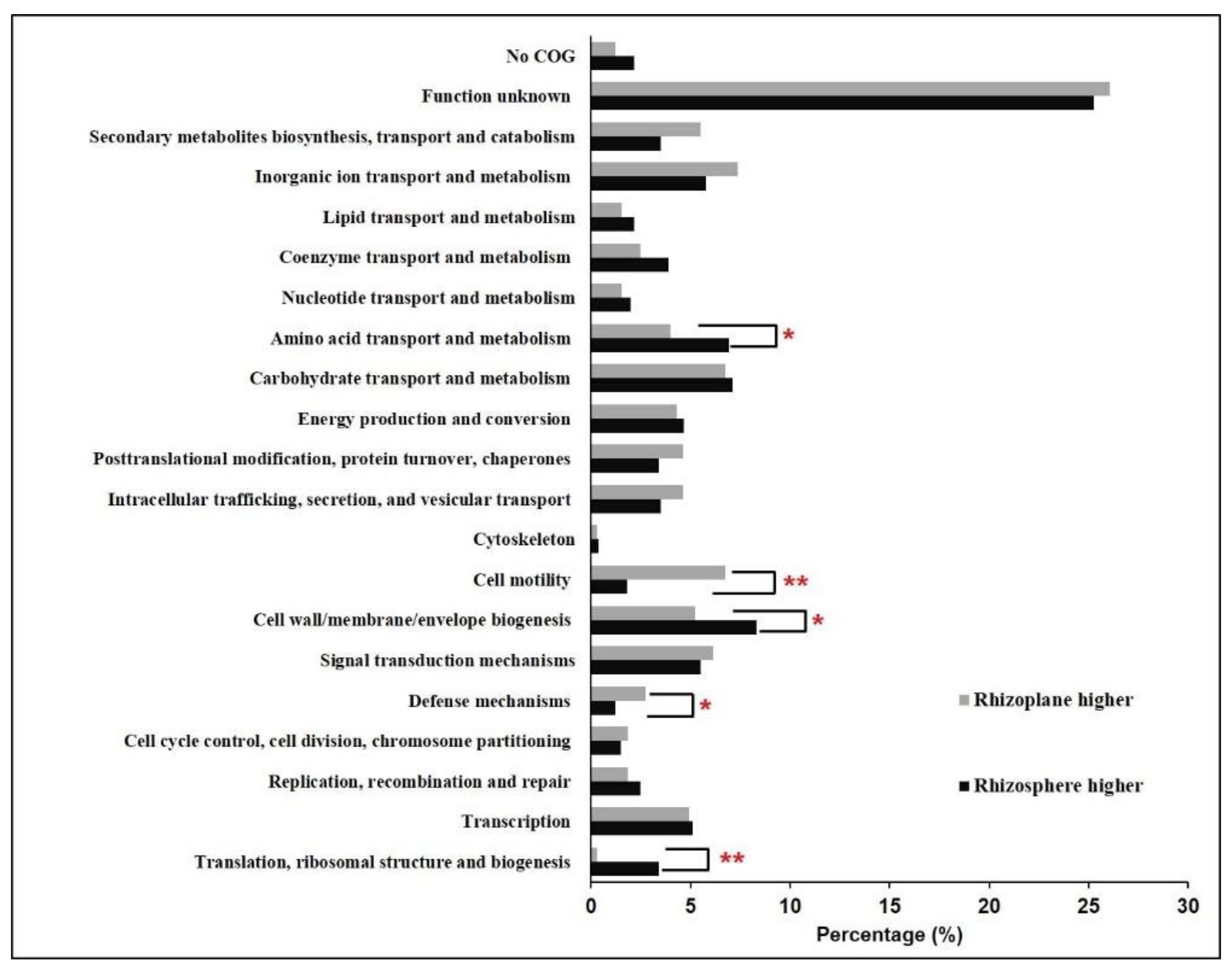
3.3. Gene Expression Comparison of Bin79 between the Rhizosphere and Rhizoplane Niches
3.4. Influence of HGT on the Citrus Rhizoplane Adaptation Capability of Bin79
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deboy, R.T.; Mongodin, E.F.; Fouts, D.E.; Tailford, L.E.; Khouri, H.; Emerson, J.B.; Mohamoud, Y.; Watkins, K.; Henrissat, B.; Gilbert, H.J.; et al. Insights into plant cell wall degradation from the genome sequence of the soil bacterium Cellvibrio japonicus. J. Bacteriol. 2008, 190, 5455–5463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofek-Lalzar, M.; Sela, N.; Goldman-Voronov, M.; Green, S.J.; Hadar, Y.; Minz, D. Niche and host-associated functional signatures of the root surface microbiome. Nat. Commun. 2014, 5, 4950. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Lin, B.; Yu, Y. Draft genome sequence of a Xylanase-producing bacterial strain, Cellvibrio mixtus J3-8. Genome Announc. 2014, 2, e01281-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syazni, M.; Yanagisawa, M.; Kasuu, M.; Nakasaki, K.; Ariga, O. Draft genome sequence of the nonmarine agarolytic bacterium Cellvibrio sp. OA-2007. Microbiol. Resour. Ann. 2015, 3, e00468-15. [Google Scholar]
- Xie, Z.; Lin, W.; Luo, J. Genome sequence of Cellvibrio pealriver PR1, a xylanolytic and agarolytic bacterium isolated from freshwater. J. Biotechnol. 2015, 214, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Riera, N.; Jin, T.; Li, J.; Wang, N. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 2017, 5, 97. [Google Scholar] [CrossRef]
- Gardner, J.G. Polysaccharide degradation systems of the saprophytic bacterium Cellvibrio japonicus. World J. Microbiol. Biotechnol. 2016, 32, 121. [Google Scholar] [CrossRef]
- Reinhold-Hurek, B.; Bunger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome: Global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Kikot, G.E.; Hours, R.A.; Alconada, T.M. Contribution of cell wall degrading enzymes to pathogenesis of Fusarium graminearum: A review. J. Basic Microb. 2009, 49, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Vicre-Gibouin, M.; Farrant, J.M.; Driouich, A. Adaptations of higher plant cell walls to water loss: Drought vs desiccation. Physiol. Plant 2008, 134, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Savatin, D.V.; Sicilia, F.; Gramegna, G.; Cervone, F.; Lorenzo, G.D. Oligogalacturonides: Plant damage-associated molecular patterns and regulators of growth and development. Front. Plant. Sci. 2013, 4, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassani, M.A.; Duran, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Zhang, P.; Trivedi, P.; Riera, N.; Wang, Y.; Liu, X.; Fan, G.; Tang, J.; Coletta-Filho, H.D.; et al. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Wu, M.; Scott, A.J. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 2012, 28, 1033–1034. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lio, P.; Crescenzi, P.; Fani, R.; Fondi, M. MEDUSA: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. 2017. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 6 February 2017).
- Ha, S.M.; Kim, C.K.; Roh, J.; Byun, J.H.; Yang, S.J.; Choi, S.B.; Chun, J.; Yong, D. Application of the whole genome-based bacterial identification system, TrueBac ID, using clinical isolates that were not identified with three Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) systems. Ann. Lab. Med. 2019, 39, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Env. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Cohen, O.; Ashkenazy, H.; Belinky, F.; Huchon, D.; Pupko, T. GLOOME: Gain loss mapping engine. Bioinformatics 2010, 26, 2914–2915. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Interpolated variable order motifs for identification of horizontally acquired DNA: Revisiting the Salmonella pathogenicity islands. Bioinformatics 2006, 22, 2196–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Alneberg, J.; Karlsson, C.M.G.; Divne, A.M.; Bergin, C.; Homa, F.; Lindh, M.V.; Hugerth, L.W.; Ettema, T.J.G.; Bertilsson, S.; Andersson, A.F.; et al. Genomes from uncultivated prokaryotes: A comparison of metagenome-assembled and single-amplified genomes. Microbiome 2018, 6, 173. [Google Scholar] [CrossRef] [Green Version]
- Galambos, D.; Anderson, R.E.; Reveillaud, J.; Huber, J.A. Genome-resolved metagenomics and metatranscriptomics reveal niche differentiation in functionally redundant microbial communities at deep-sea hydrothermal vents. Env. Microbiol. 2019, 21, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Walker, A.W.; Roehe, R.; Watson, M. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953. [Google Scholar] [CrossRef] [Green Version]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Dick, G.J.; Andersson, A.F.; Baker, B.J.; Simmons, S.L.; Yelton, A.P.; Banfield, J.F. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009, 10, R85. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.R.; He, J.Z. Characterization of a xylanase-producing Cellvibrio mixtus strain J3-8 and its genome analysis. Sci. Rep. 2015, 5, 10521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, J.G.; Keating, D.H. Requirement of the type II secretion system for utilization of cellulosic substrates by Cellvibrio japonicus. Appl. Env. Microbiol. 2010, 76, 5079–5087. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Suzuki, H.; Hirotani, M.; Ozawa, H.; Nisizawa, K. Effect of nature and supply of carbon sources on cellulase formation in Pseudomonas fluorescens var. cellulosa. J. Biochem. 1970, 67, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Monge, E.C.; Tuveng, T.R.; Vaaje-Kolstad, G.; Eijsink, V.G.H.; Gardner, J.G. Systems analysis of the glycoside hydrolase family 18 enzymes from Cellvibrio japonicus characterizes essential chitin degradation functions. J. Biol. Chem. 2018, 293, 3849–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, J.P.; Falomir, M.P.; Gozalbo, D. Chitin: A structural biopolysaccharide with multiple applications. In eLS; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Carrion, V.J.; Perez-Jaramillo, J.; Cordovez, V.; Tracanna, V.; de Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; van Ijcken, W.F.J.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Veliz, E.A.; Martínez-Hidalgo, P.; Hirsch, A.M. Chitinase-producing bacteria and their role in biocontrol. AIMS Microbiol. 2017, 3, 689–705. [Google Scholar] [CrossRef]
- Patten, C.L.; Glick, B.R. Bacterial biosynthesis of indole-3-acetic acid. Can. J. Microbiol. 1996, 42, 207–220. [Google Scholar] [CrossRef]
- Soutourina, O.A.; Bertin, P.N. Regulation cascade of flagellar expression in Gram-negative bacteria. FEMS Microbiol. Rev. 2003, 27, 505–523. [Google Scholar] [CrossRef] [Green Version]
- Theodorou, M.C.; Theodorou, E.C.; Kyriakidis, D.A. Involvement of AtoSC two-component system in Escherichia coli flagellar regulon. Amino Acids 2012, 43, 833–844. [Google Scholar] [CrossRef]
- Bogino, P.C.; Oliva Mde, L.; Sorroche, F.G.; Giordano, W. The role of bacterial biofilms and surface components in plant-bacterial associations. Int. J. Mol. Sci. 2013, 14, 15838–15859. [Google Scholar] [CrossRef] [Green Version]
- Hantke, K. Dihydroxybenzolyserine—A siderophore for E. coli. FEMS Microbiol. Lett. 1990, 67, 5–8. [Google Scholar]
- Rivera, M. Bacterioferritin: Structure, dynamics, and protein-protein interactions at play in iron storage and mobilization. Acc. Chem. Res. 2017, 50, 331–340. [Google Scholar] [CrossRef]
- Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005, 56, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okusu, H.; Ma, D.; Nikaido, H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 1996, 178, 306–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolwell, G.P. Role of active oxygen species and NO in plant defence responses. Curr. Opin. Plant Biol. 1999, 2, 287–294. [Google Scholar] [CrossRef]
- Mongkolsuk, S.; Helmann, J.D. Regulation of inducible peroxide stress responses. Mol. Microbiol. 2002, 45, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, C.; Ratering, S.; Geissler-Plaum, R.; Schnell, S. Rheinheimera hassiensis sp. nov. and Rheinheimera muenzenbergensis sp. nov., two species from the rhizosphere of Hordeum secalinum. 2014, 64, 1202–1209. [Google Scholar]
- Presta, L.; Bosi, E.; Fondi, M.; Maida, I.; Perrin, E.; Miceli, E.; Maggini, V.; Bogani, P.; Firenzuoli, F.; Di Pilato, V.; et al. Phenotypic and genomic characterization of the antimicrobial producer Rheinheimera sp. EpRS3 isolated from the medicinal plant Echinacea purpurea: Insights into its biotechnological relevance. Res. Microbiol. 2017, 168, 293–305. [Google Scholar] [CrossRef]
- Goldenfeld, N.; Woese, C. Biology’s next revolution. Nature 2007, 445, 369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Yang, D.; Kendall, J.R.; Borriss, R.; Druzhinina, I.S.; Kubicek, C.P.; Shen, Q.; Zhang, R. Comparative genomic analysis of Bacillus amyloliquefaciens and Bacillus subtilis reveals evolutional traits for adaptation to plant-associated habitats. Front. Microbiol. 2016, 7, 2039. [Google Scholar] [CrossRef]
- Abdian, P.L.; Caramelo, J.J.; Ausmees, N.; Zorreguieta, A. RapA2 is a Calcium-binding lectin composed of two highly conserved Cadherin-like domains that specifically recognize Rhizobium leguminosarum acidic exopolysaccharides. J. Biol. Chem. 2013, 288, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, N.; Skurray, R. Characterization of the F plasmid bifunctional conjugation gene, traG. Mol. Gen. Genet. 1992, 232, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Solden, L.; Lloyd, K.; Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Value |
|---|---|
| Total reading base pairs (bp) | 5713,429 |
| Scaffold number | 11 |
| N50 (bp) | 1795,691 |
| Read coverage | 15.68 |
| GC content (%) | 46.21 |
| tRNAs | 36 |
| Protein-coding sequences | 4754 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Xu, J.; Wang, E.; Wang, N. Mechanisms Underlying the Rhizosphere-To-Rhizoplane Enrichment of Cellvibrio Unveiled by Genome-Centric Metagenomics and Metatranscriptomics. Microorganisms 2020, 8, 583. https://doi.org/10.3390/microorganisms8040583
Zhang Y, Xu J, Wang E, Wang N. Mechanisms Underlying the Rhizosphere-To-Rhizoplane Enrichment of Cellvibrio Unveiled by Genome-Centric Metagenomics and Metatranscriptomics. Microorganisms. 2020; 8(4):583. https://doi.org/10.3390/microorganisms8040583
Chicago/Turabian StyleZhang, Yunzeng, Jin Xu, Entao Wang, and Nian Wang. 2020. "Mechanisms Underlying the Rhizosphere-To-Rhizoplane Enrichment of Cellvibrio Unveiled by Genome-Centric Metagenomics and Metatranscriptomics" Microorganisms 8, no. 4: 583. https://doi.org/10.3390/microorganisms8040583