Microbial Metabolic Potential of Phenol Degradation in Wastewater Treatment Plant of Crude Oil Refinery: Analysis of Metagenomes and Characterization of Isolates


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Shotgun Metagenomics
2.3. Prokaryotic Taxonomic Assignment of Shotgun Metagenomics Datasets
2.4. Identifying CDS Coding for Protein Homologs of the C12O, C23O and PH Families
2.5. Isolation and Identification of Strains
2.6. Sequencing and Analysis of the Genomes of the Isolated Strains Acinetobacter venetianus ICP1 and Pseudomonas oleovorans ICTN13
2.7. Cultivation of Bacteria on Phenol Liquid Medium
2.8. Chemical Analyses
2.9. Determination of Specific Activities of Enzymes
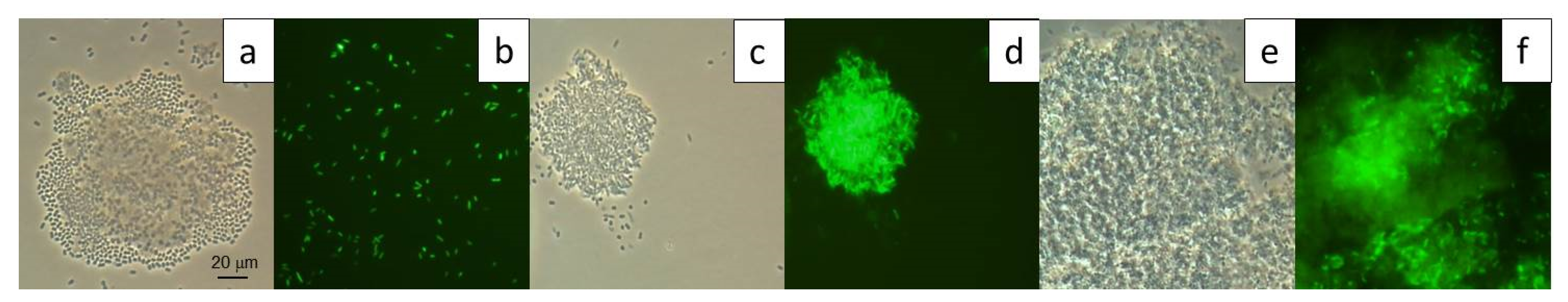
2.10. GFP-Tagging of Strains ICTN13 and ICP1
2.11. Flow Cytometry Analysis
2.12. Effect of Higher Phenol Concentrations on the Growth of the Strains
2.13. Biofilm Formation Assay in Multi-Well Plates
2.14. Data Availability
3. Results and Discussion
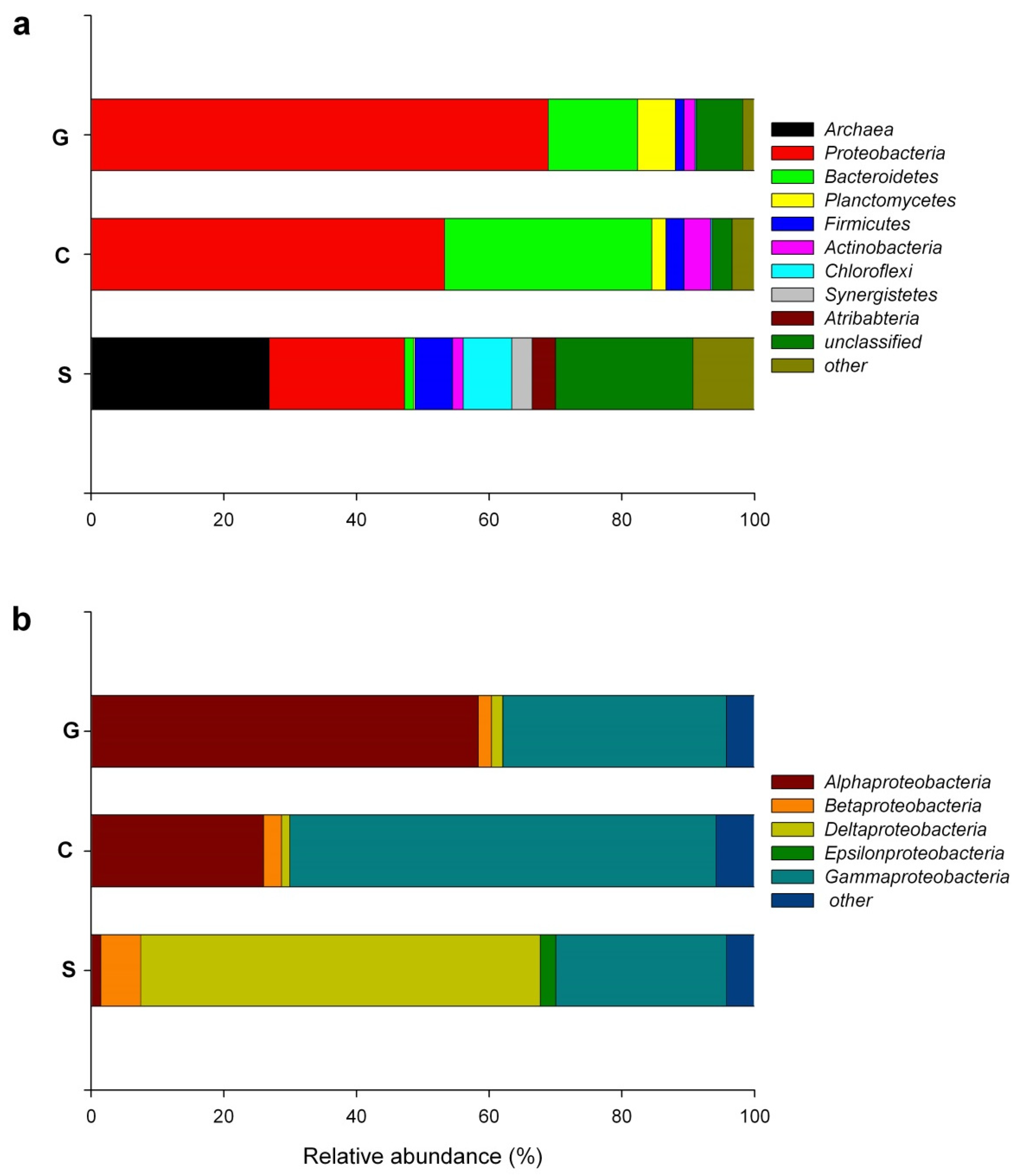
3.1. Changes in Composition of the Bacterial Communities in the Different Steps of the Crude Oil Refinery WWTP
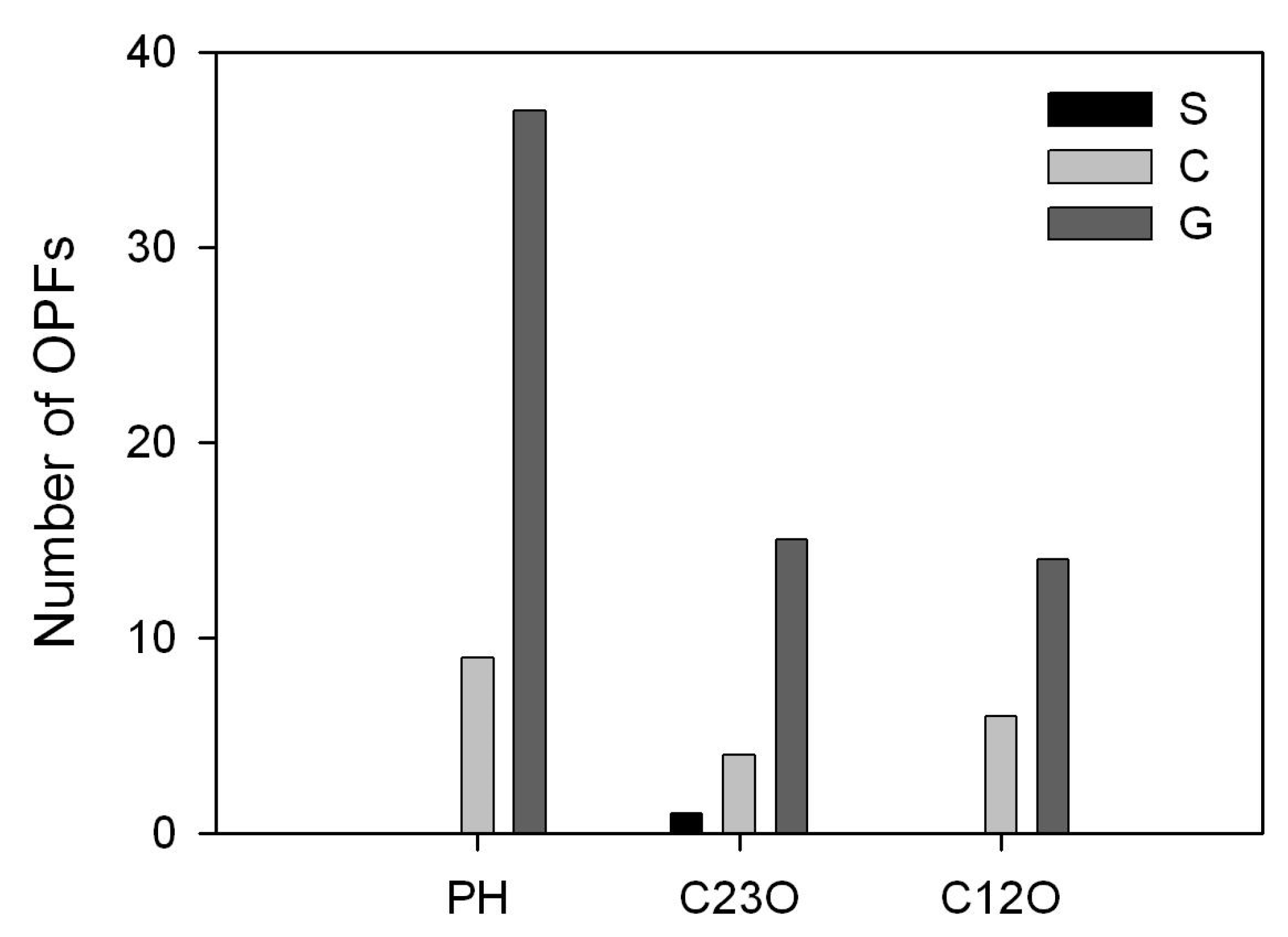
3.2. Diversity of Key Catabolic Genes (PH, C12O and C23O) of Phenol Degradation in the WWTP Samples
3.3. Diversity of Aromatic Compounds Degrading Isolates
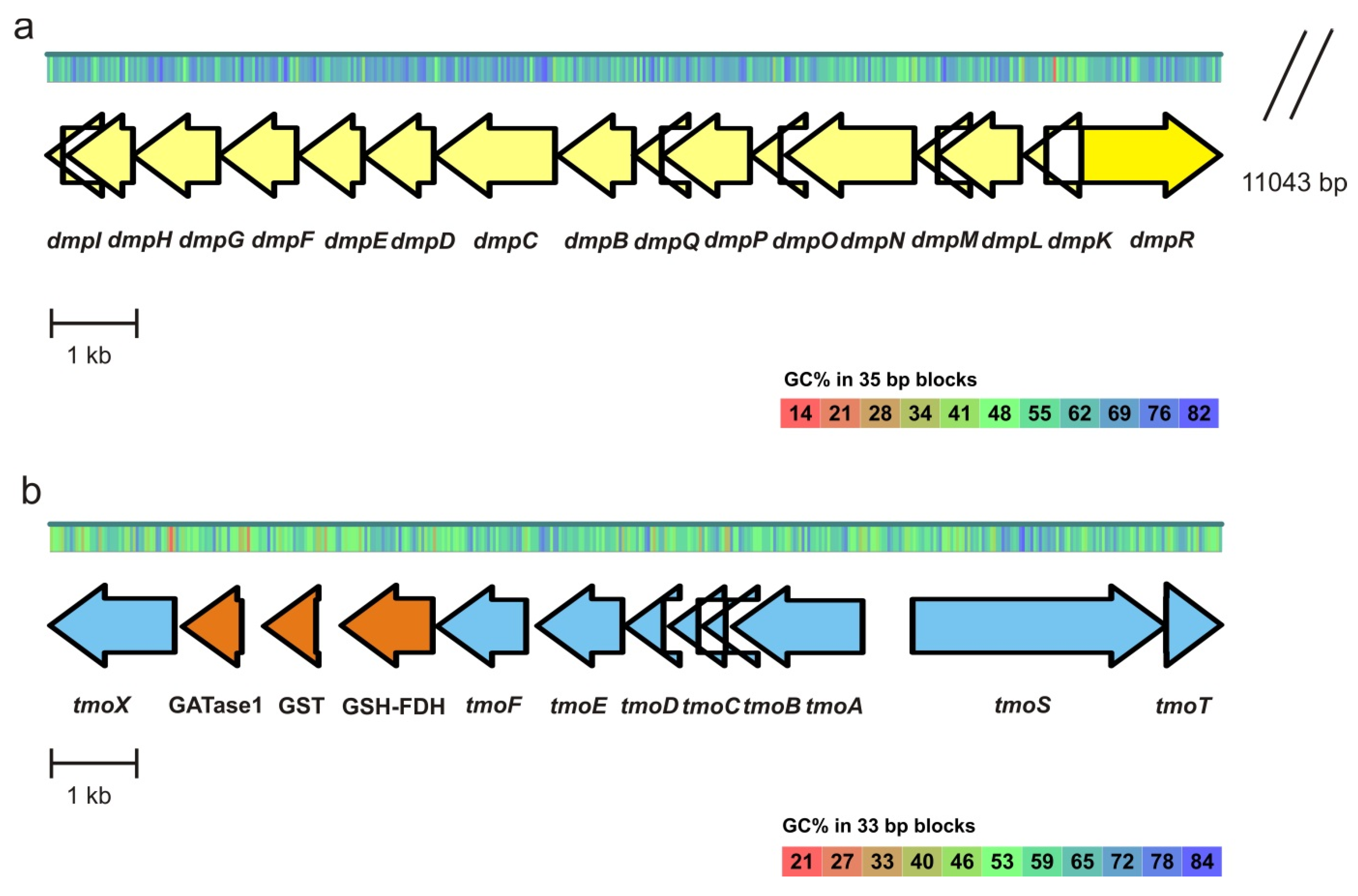
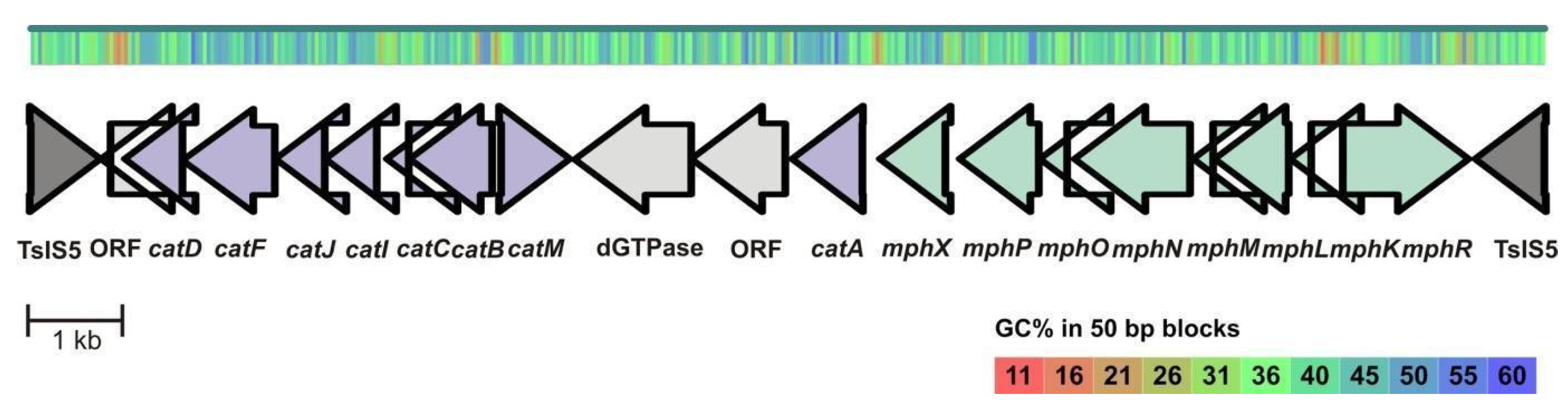
3.4. Analysis of Genes of the Aromatic Compounds Degradation Pathways of the Strains ICTN13 and ICP1
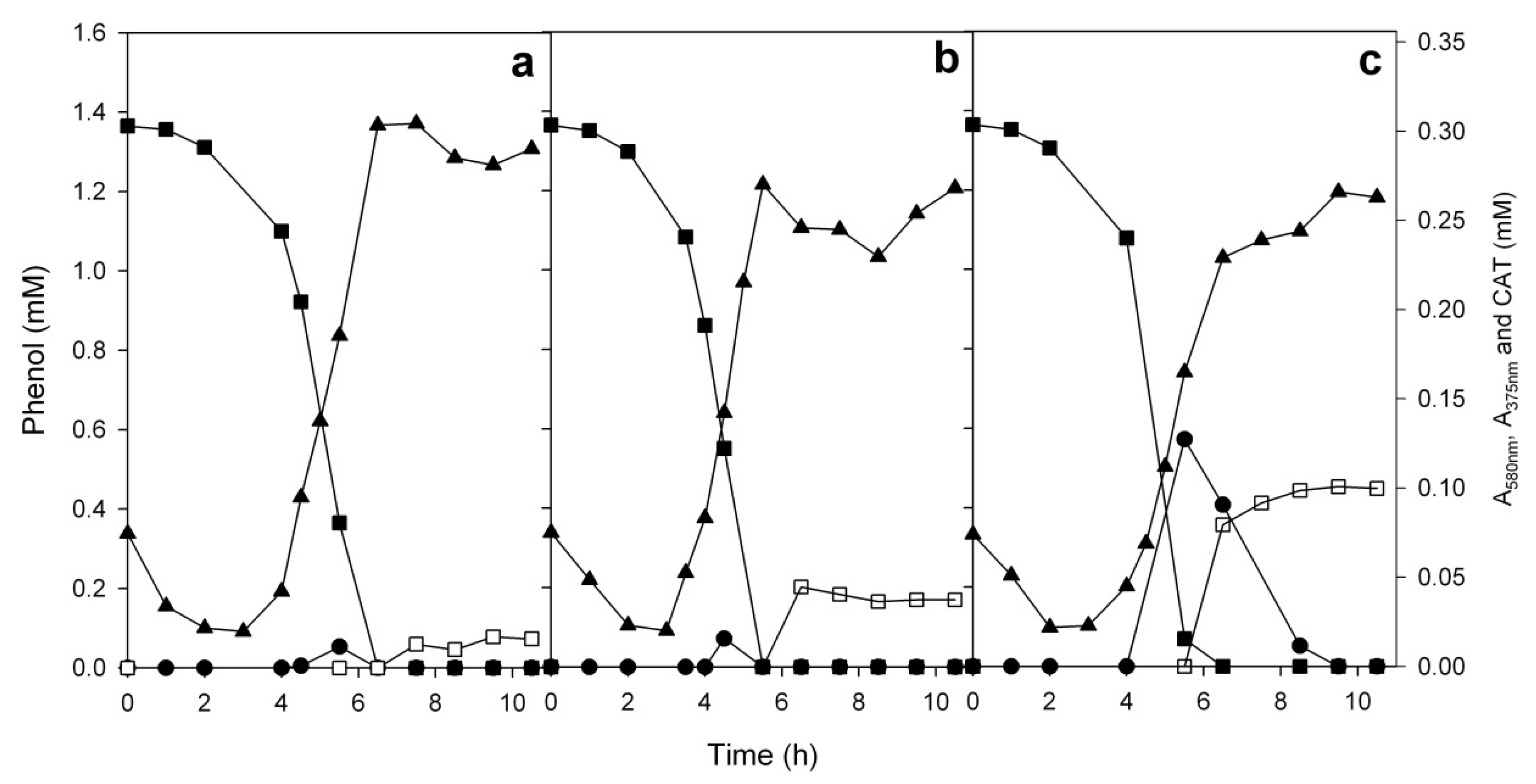
3.5. Comparison of the Growth of the Strains ICP1 and ICTN13 on Phenol
3.6. Growth of the Mixed Culture of the Strains ICP1 and ICTN13 on Phenol
3.7. Effect of Higher Phenol Concentrations on the Growth of the Strains ICP1 and ICTN13
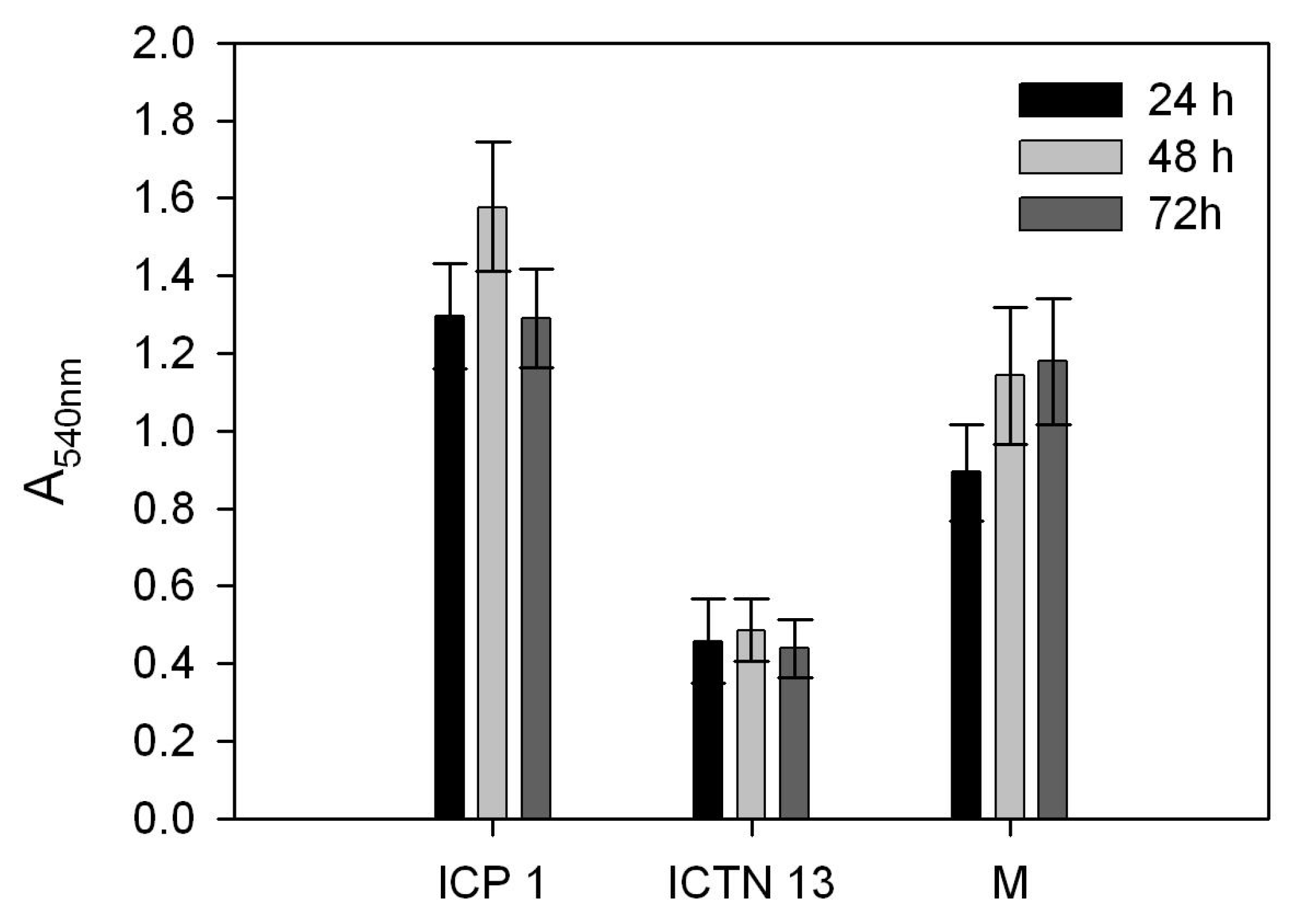
3.8. Comparison of Biofilm Formation Ability of the Strains ICP1 and ICTN13, and Their Mixture
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vendramel, S.; Bassin, J.P.; Dezotti, M.; Sant’Anna, G.L., Jr. Treatment of petroleum refinery wastewater containing heavily polluting substances in an aerobic submerged fixed-bed reactor. Environ. Technol. 2015, 36, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Banat, F.; Almansoori, A.; Haija, M.A. Review of technologies for biotreatment of refinery wastewaters: Progress, challenges and future opportunities. Environ. Technol. Rev. 2016, 5, 12–38. [Google Scholar] [CrossRef]
- Saien, J.; Nejati, H. Enhanced photocatalytic degradation of pollutants in petroleum refinery wastewater under mild conditions. J. Hazard. Mater. 2007, 148, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Ahmadun, F.R.; Pendashteh, A.; Abdullah, L.C.; Biak, D.R.A.; Madaeni, S.S.; Abidin, Z.Z. Review of technologies for oil and gas produced water treatment. J. Hazard. Mater. 2009, 170, 530–551. [Google Scholar]
- Ashfaq, A.; Saadia, A.; Anees, S.; Khursheed, A. Environmental waste management: A case study of refinery. J. Ind. Pollut. Control. 2010, 26, 95–103. [Google Scholar]
- Sarkar, J.; Kazy, S.K.; Gupta, A.; Dutta, A.; Mohapatra, B.; Roy, A.; Bera, P.; Mitre, A.; Sari, P. Biostimulation of indigenous microbial community for bioremediation of petroleum refinery sludge. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Head, I.M.; Gray, N.D.; Larter, S.R. Life in the slow lane; biogeochemistry of biodegraded petroleum containing reservoirs and implications for energy recovery and carbon management. Front. Microbiol. 2014, 5, 566. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Gutierrez, J.; Figueras, A.; Albaiges, J.; Jimenez, N.; Vinas, M.; Solanas, A.M.; Novoa, B. Bacterial communities from shoreline environments (Costa da Morte, Northwestern Spain) affected by the Prestige oil spill. Appl. Environ. Microbiol. 2009, 75, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Neethu, C.S.; Saravanakumar, C.; Purvaja, R.; Robin, R.S.; Ramesh, R. Oil-spill triggered shift in indigenous microbial structure and functional dynamics in different marine environmental matrices. Sci. Rep. 2019, 9, 1354. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.; Kargari, A.; Sanaeepur, H.; Abbassian, K.; Najafi, A.; Mofarrah, E. Phenol removal from industrial wastewaters: A short review. Desalin. Water Treat. 2015, 53, 2215–2234. [Google Scholar] [CrossRef]
- Al-Khalid, T.; El-Naas, M.H. Organic Contaminants in Refinery Wastewater: Characterization and Novel Approaches for Biotreatment. In Recent Insights in Petroleum Science and Engineering; Zoveidavianpoor, M., Ed.; eBook; InTech: Rijeka, Croatia, 2017. [Google Scholar] [CrossRef]
- Liu, J.Q.; Bacosa, H.P.; Liu, Z.F. Potential environmental factors affecting oil-degrading bacterial populations in deep and surface waters of the northern Gulf of Mexico. Front. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson-Palme, J.; Hartmann, M.; Eriksson, K.M.; Pal, C.; Thorell, K.; Larsson, D.G.; Nilsson, R.H. METAXA2: Improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 2015, 15, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [Green Version]
- Yakimov, M.M.; Golyshin, P.N.; Lang, S.; Moore, E.R.B.; Abraham, W.R.; Lunsdorf, H.; Timmis, K.N. Alcanivorax borkumensis gen. nov., sp. nov., a new, hydrocarbon-degrading and surfactant-producing marine bacterium. Int. J. Syst. Bacteriol. 1998, 48, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Huu, N.B.; Denner, E.B.M.; Ha, D.T.C.; Wanner, G.; Stan-Lotter, H. Marinobacter aquaeolei sp. nov., a halophilic bacterium isolated from a Vietnamese oil-producing well. Int. J. Syst. Bacteriol. 1999, 49, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Viggor, S.; Heinaru, E.; Künnapas, A.; Heinaru, A. Evaluation of different phenol hydroxylase-possessing phenol-degrading pseudomonads by kinetic parameters. Biodegradation 2008, 19, 759–769. [Google Scholar] [CrossRef]
- Jõesaar, M.; Viggor, S.; Heinaru, E.; Naanuri, E.; Mehike, M.; Leito, I.; Heinaru, A. Strategy of Pseudomonas pseudoalcaligenes C70 for effective degradation of phenol and salicylate. PLoS ONE 2017, 12, e0173180. [Google Scholar] [CrossRef] [PubMed]
- Hegeman, G.D. Synthesis of the enzymes of the mandelate pathway by Pseudomonas putida. I. Synthesis of enzymes by the wild type. J. Bacteriol. 1966, 91, 1140–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Koch, B.; Jensen, L.E.; Nybroe, O. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. J. Microbiol. Methods 2001, 45, 187–195. [Google Scholar] [CrossRef]
- Lambertsen, L.; Sternberg, C.; Molin, S. Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ. Microbiol. 2004, 6, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, A. The effects of culture concentration and age, time and temperature on bacterial attachments to polystyrene. Can. J. Microbiol. 1977, 23, 1–6. [Google Scholar] [CrossRef]
- Jakovleva, J.; Teppo, A.; Velts, A.; Saumaa, S.; Moor, H.; Kivisaar, M.; Teras, R. Fis regulates the competitiveness of Pseudomonas putida on barley roots by inducing biofilm formation. Microbiology 2012, 158, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Soares-Castro, P.; Yadav, T.C.; Viggor, S.; Kivisaar, M.; Kapley, A.; Santos, P.M. Seasonal bacterial community dynamics in a crude oil refinery wastewater treatment plant. Appl. Microbiol. Biotechnol. 2019, 103, 9131–9141. [Google Scholar] [CrossRef]
- Tan, B.; Nesbo, C.; Foght, J. Re-analysis of omics data indicates Smithella may degrade alkanes by addition to fumarate under methanogenic conditions. Isme J. 2014, 8, 2353–2356. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.L.; Hanada, S.; Ohashi, A.; Harada, H.; Kamagata, Y.; Sekiguchi, Y. Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl. Environ. Microbiol. 2008, 74, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, T.; Aitken, M.D. Role of methylotrophs in the degradation of hydrocarbons during the Deepwater Horizon oil spill. Isme J. 2014, 8, 2543–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, R. Marinobacter; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Engel, A.S.; Liu, C.; Paterson, A.T.; Anderson, L.C.; Turner, R.E.; Overton, E.B. Salt Marsh Bacterial Communities before and after the Deepwater Horizon Oil Spill. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahi, A.; Aydin, S.; Ince, B.; Ince, O. Reconstruction of bacterial community structure and variation for enhanced petroleum hydrocarbons degradation through biostimulation of oil contaminated soil. Chem. Eng. J. 2016, 306, 60–66. [Google Scholar] [CrossRef]
- Acosta-Gonzalez, A.; Marques, S. Bacterial diversity in oil-polluted marine coastal sediments. Curr. Opin. Biotechnol. 2016, 38, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, E.A.; Conrad, M.E.; Chakraborty, R.; Bill, M.; Borglin, S.E.; Hollibaugh, J.T.; Mason, O.U.; Y, M.P.; Reid, F.C.; Stringfellow, W.T.; et al. Succession of hydrocarbon-degrading bacteria in the aftermath of the deepwater horizon oil spill in the gulf of Mexico. Environ. Sci. Technol. 2013, 47, 10860–10867. [Google Scholar] [CrossRef]
- Lamendella, R.; Strutt, S.; Borglin, S.; Chakraborty, R.; Tas, N.; Mason, O.U.; Hultman, J.; Prestat, E.; Hazen, T.C.; Jansson, J.K. Assessment of the Deepwater Horizon oil spill impact on Gulf coast microbial communities. Front. Microbiol. 2014, 5, 130. [Google Scholar] [CrossRef] [Green Version]
- Kostka, J.E.; Prakash, O.; Overholt, W.A.; Green, S.J.; Freyer, G.; Canion, A.; Delgardio, J.; Norton, N.; Hazen, T.C.; Huettel, M. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 2011, 77, 7962–7974. [Google Scholar] [CrossRef] [Green Version]
- Beazley, M.J.; Martinez, R.J.; Rajan, S.; Powell, J.; Piceno, Y.M.; Tom, L.M.; Andersen, G.L.; Hazen, T.C.; Van Nostrand, J.D.; Zhou, J.; et al. Microbial community analysis of a coastal salt marsh affected by the Deepwater Horizon oil spill. PLoS ONE 2012, 7, e41305. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J. Evaluating bacterial community structures in oil collected from the sea surface and sediment in the northern Gulf of Mexico after the Deepwater Horizon oil spill. Microbiol. Open 2013, 2, 492–504. [Google Scholar] [CrossRef]
- Antoniou, E.; Fodelianakis, S.; Korkakaki, E.; Kalogerakis, N. Biosurfactant production from marine hydrocarbon-degrading consortia and pure bacterial strains using crude oil as carbon source. Front. Microbiol. 2015, 6, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, B.G.; Singh, H.; Bhatnagar, A.K.; Sinha, A.K.; Singh, R.R. Petroleum systems of the Mumbai offshore basin, India. In Proceedings of the AAPG Annual Convention, Long Beach, CA, USA, 1–4 April 2007; AAPG Search and Discovery. AAPG: Tulsa, OK, USA, 2007. Article 10154. [Google Scholar]
- Bugg, T.D.; Ramaswamy, S. Non-heme iron-dependent dioxygenases: Unravelling catalytic mechanisms for complex enzymatic oxidations. Curr. Opin. Chem. Biol. 2008, 12, 134–140. [Google Scholar] [CrossRef]
- Shukla, A.; Singh, B.; Cameotra, S.S.; Kahlon, R.S. Pseudomonas Oxygenases: Nature and Function. In Pseudomonas: Molecular and Applied Biology; Kahlon, R.S., Ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Nie, Y.; Tang, Y.Q.; Li, Y.; Chi, C.Q.; Cai, M.; Wu, X.L. The genome sequence of Polymorphum gilvum SL003B-26A1T reveals its genetic basis for crude oil degradation and adaptation to the saline soil. PLoS ONE 2012, 7, e0031261. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Q.; Xu, L.; Liu, X.Y.; Zhao, G.F.; Cai, H.; Nie, Y.; Wu, X.L. Functional genetic diversity and culturability of petroleum-degrading bacteria isolated from oil-contaminated soils. Front. Microbiol. 2018, 9, 1332. [Google Scholar] [CrossRef]
- Stefani, F.O.P.; Bell, T.H.; Marchand, C.; de la Providencia, I.E.; El Yassimi, A.; St-Arnaud, M.; Hijri, M. Culture-dependent and -independent methods capture different microbial community fractions in hydrocarbon-contaminated soils. PLoS ONE 2015, 10, e0128272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khalid, T.; El-Naas, M.H. Aerobic biodegradation of phenols: A comprehensive review. Crit. Rev. Environ. Sci. Tec. 2012, 42, 1631–1690. [Google Scholar] [CrossRef]
- Powlowski, J.; Shingler, V. Genetic and biochemistry of phenol degradation by Pseudomonas sp. CF600. Biodegradation 1994, 5, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gonzalez, M.I.; Olson, M.; Gatenby, A.A.; Mosqueda, G.; Manzanera, M.; Campos, M.J.; Vichez, S.; Ramos, J.L. Cross-regulation between a novel two-component signal transduction system for catabolism of toluene in Pseudomonas mendocina and the TodST system from Pseudomonas putida. J. Bacteriol. 2002, 184, 7062–7067. [Google Scholar] [CrossRef] [Green Version]
- Parales, R.E.; Parales, J.V.; Pelletier, D.A.; Ditty, J.L. Diversity of microbial toluene degradation pathways. Adv. Appl. Microbiol. 2008, 64, 1–73. [Google Scholar]
- Nishino, S.F.; Spain, J.C.; Belcher, L.A.; Litchfield, C.D. Chlorobenzene degradation by bacteria isolated from contaminated groundwater. Appl. Environ. Microbiol. 1992, 58, 1719–1726. [Google Scholar] [CrossRef] [Green Version]
- Santos, P.M.; Mignogna, G.; Heipieper, H.J.; Zennaro, E. Occurrence and properties of glutathione s-transferases in phenol-degrading Pseudomonas strains. Res. Microbiol. 2002, 153, 89–98. [Google Scholar] [CrossRef]
- Allocati, N.; Federici, L.; Masulli, M.; Di Ilio, C. Glutathione transferases in bacteria. Febs J. 2009, 276, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Cafaro, V.; Izzo, V.; Scognamiglio, R.; Notomista, E.; Capasso, P.; Casbarra, A.; Pucci, P.; Di Donato, A. Phenol hydroxylase and toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1: Interplay between two enzymes. Appl. Environ. Microbiol. 2004, 70, 2211–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafaro, V.; Notomista, E.; Capasso, P.; Di Donato, A. Regiospecificity of two multicomponent monooxygenases from Pseudomonas stutzeri OX1: Molecular basis for catabolic adaptation of this microorganism to methylated aromatic compounds. Appl. Environ. Microbiol. 2005, 71, 4736–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenghi, F.L.; Berlanda, D.; Galli, E.; Sello, G.; Barbieri, P. Organization and regulation of meta cleavage pathway genes for toluene and o-xylene derivative degradation in Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 2001, 67, 3304–3308. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.H.; Studts, J.M.; Fox, B.G. Combined participation of hydroxylase active site residues and effector protein binding in a para to ortho modulation of toluene 4-monooxygenase regiospecificity. Biochemistry 2002, 41, 3176–3188. [Google Scholar] [CrossRef]
- McClay, K.; Fox, B.G.; Steffan, R.J. Toluene monooxygenase-catalyzed epoxidation of alkenes. Appl. Environ. Microbiol. 2000, 66, 1877–1882. [Google Scholar] [CrossRef] [Green Version]
- Bertoni, G.; Bolognese, F.; Galli, E.; Barbieri, P. Cloning of the genes for and characterization of the early stages of toluene and o-xylene catabolism in Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 1996, 62, 3704–3711. [Google Scholar] [CrossRef] [Green Version]
- Sazinsky, M.H.; Bard, J.; Di Donato, A.; Lippard, S.J. Crystal structure of the toluene/o-xylene monooxygenase hydroxylase from Pseudomonas stutzeri OX1. Insight into the substrate specificity, substrate channeling, and active site tuning of multicomponent monooxygenases. J. Biol. Chem. 2004, 279, 30600–30610. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Peng, Z.; Zhan, Y.; Wang, J.; Yan, Y.; Chen, M.; Lu, W.; Ping, S.; Zhang, W.; Zhao, Z.; et al. Novel regulator MphX represses activation of phenol hydroxylase genes caused by a XylR/DmpR-type regulator MphR in Acinetobacter calcoaceticus. PLoS ONE 2011, 6, e17350. [Google Scholar] [CrossRef]
- Zhan, Y.; Yan, Y.; Zhang, W.; Chen, M.; Lu, W.; Ping, S.; Lin, M. Comparative analysis of the complete genome of an Acinetobacter calcoaceticus strain adapted to a phenol-polluted environment. Res. Microbiol. 2012, 163, 36–43. [Google Scholar] [CrossRef]
- Diaz, E.; Prieto, M.A. Bacterial promoters triggering biodegradation of aromatic pollutants. Curr. Opin. Biotechnol. 2000, 11, 467–475. [Google Scholar] [CrossRef]
- Tropel, D.; van der Meer, J.R. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol. Mol. Biol. Rev. 2004, 68, 474–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Dal, S.; Trautwein, G.; Gerischer, U. Transcriptional organization of genes for protocatechuate and quinate degradation from Acinetobacter sp. strain ADP1. Appl. Environ. Microbiol. 2005, 71, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siehler, S.Y.; Dal, S.; Fischer, R.; Patz, P.; Gerischer, U. Multiple-level regulation of genes for protocatechuate degradation in Acinetobacter baylyi includes cross-regulation. Appl. Environ. Microbiol. 2007, 73, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tian, Y.; Han, B.; Zhao, H.B.; Bi, J.N.; Cai, B.L. Biodegradation of phenol by free and immobilized Acinetobacter sp. strain PD12. J. Environ. Sci.-China 2007, 19, 222–225. [Google Scholar] [CrossRef]
- Obach, R.S.; Kalgutkar, A.S. Reactive Electrophiles and Metabolic Activation. In Comprehensive Toxicology, Vol 1: General Principles, 2nd ed.; McQueen, C.A., Ed.; Elsevier Ltd.: Kidlington, UK, 2010; pp. 309–347. [Google Scholar]
- El-Naas, M.H.; Al-Muhtaseb, S.A.; Makhlouf, S. Biodegradation of phenol by Pseudomonas putida immobilized in polyvinyl alcohol (PVA) gel. J. Hazard. Mater. 2009, 164, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Hosein, S.G.; Millette, D.; Butler, B.J.; Greer, C.W. Catabolic gene probe analysis of an aquifer microbial community degrading creosote-related polycyclic aromatic and heterocyclic compounds. Microb. Ecol. 1997, 34, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Heinaru, E.; Merimaa, M.; Viggor, S.; Lehiste, M.; Leito, I.; Truu, J.; Heinaru, A. Biodegradation efficiency of functionally important populations selected for bioaugmentation in phenol- and oil-polluted area. FEMS Microbiol. Ecol. 2005, 51, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.A.; Syed, M.A.; Arif, N.M.; Shukor, M.Y.A.; Shamaan, N.A. Isolation, identification and characterization of elevated phenol degrading Acinetobacter sp. strain AQ5NOL 1. Aust. J. Basic Appl. Sci. 2011, 5, 1035–1045. [Google Scholar]
- Cases, I.; de Lorenzo, V. Promoters in the environment: Transcriptional regulation in its natural context. Nat. Rev. Microbiol. 2005, 3, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Lin, J.; Zhao, J.; Xu, T.; Yang, Q.; Zhao, J.; Zhao, Z.; Song, X. Changes in the microbial community diversity of oil exploitation. Genes 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ławniczak, Ł.; Woźniak-Karczewska, M.; Loibner, A.P.; Heipieper, H.J.; Chrzanowski, Ł. Microbial degradation of hydrocarbons—Basic principles for bioremediation: A review. Molecules 2020, 25, 856. [Google Scholar] [CrossRef] [Green Version]
- Molin, G.; Nilsson, I. Degradation of phenol by Pseudomonas putida ATCC-11172 in continuous culture at different ratios of biofilm surface to culture volume. Appl. Environ. Microbiol. 1985, 50, 946–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.L.; Tay, J.H.; Tay, S.T.L. Aggregation of immobilized activated sludge cells into aerobically grown microbial granules for the aerobic biodegradation of phenol. Lett. Appl. Microbiol. 2002, 35, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Pourakbar, M.; Moussavi, G.; Yaghmaeian, K. Enhanced biodegradation of phenol in a novel cyclic activated sludge integrated with a rotating bed bioreactor in anoxic and peroxidase-mediated conditions. Rsc. Adv. 2018, 8, 6293–6305. [Google Scholar] [CrossRef] [Green Version]
- Hori, K.; Yamashita, S.; Ishii, S.; Kitagawa, M.; Tanji, Y.; Unno, H. Isolation, characterization and application to off-gas treatment of toluene-degrading bacteria. J. Chem. Eng. Jpn. 2001, 34, 1120–1126. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Koki, J.; Unno, H.; Hori, K. Two morphological types of cell appendages on a strongly adhesive bacterium, Acinetobacter sp. strain Tol 5. Appl. Environ. Microbiol. 2004, 70, 5026–5029. [Google Scholar] [CrossRef] [Green Version]
- Adav, S.S.; Chen, M.Y.; Lee, D.J.; Ren, N.Q. Degradation of phenol by Acinetobacter strain isolated from aerobic granules. Chemosphere 2007, 67, 1566–1572. [Google Scholar] [CrossRef]
- Simões, L.C.; Simões, M.; Vieira, M.J. Intergeneric coaggregation among drinking water bacteria: Evidence of a role for Acinetobacter calcoaceticus as a bridging bacterium. Appl. Environ. Microbiol. 2008, 74, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Phuong, K.; Kakii, K.; Nikata, T. Intergeneric coaggregation of non-flocculating Acinetobacter spp. isolates with other sludge-constituting bacteria. J. Biosci. Bioeng. 2009, 107, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; Dorsey, C.W.; Edelmann, R.E.; Actis, L.A. Attachment to and biofilm formation on abiotic surfaces by Acinetobacter baumannii: Involvement of a novel chaperone-usher pili assembly system. Microbiology 2003, 149, 3473–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.; Sakamoto, M.; Ono, T.; Kakii, K. Coaggregation between Acinetobacter johnsonii S35 and Microbacterium esteraromaticum strains isolated from sewage activated sludge. J. Biosci. Bioeng. 2003, 96, 10–15. [Google Scholar] [CrossRef]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, H.; Sivaneson, M.; Filloux, A. Key two-component regulatory systems that control biofilm formation in Pseudomonas aeruginosa. Environ. Microbiol. 2011, 13, 1666–1681. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.B.; Haagensen, J.A.; Heydorn, A.; Molin, S. Metabolic commensalism and competition in a two-species microbial consortium. Appl. Environ. Microbiol. 2002, 68, 2495–2502. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.K.; Haagensen, J.A.; Gjermansen, M.; Jorgensen, T.M.; Tolker-Nielsen, T.; Molin, S. Characterization of a Pseudomonas putida rough variant evolved in a mixed-species biofilm with Acinetobacter sp. strain C6. J. Bacteriol. 2007, 189, 4932–4943. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viggor, S.; Jõesaar, M.; Soares-Castro, P.; Ilmjärv, T.; Santos, P.M.; Kapley, A.; Kivisaar, M. Microbial Metabolic Potential of Phenol Degradation in Wastewater Treatment Plant of Crude Oil Refinery: Analysis of Metagenomes and Characterization of Isolates. Microorganisms 2020, 8, 652. https://doi.org/10.3390/microorganisms8050652
Viggor S, Jõesaar M, Soares-Castro P, Ilmjärv T, Santos PM, Kapley A, Kivisaar M. Microbial Metabolic Potential of Phenol Degradation in Wastewater Treatment Plant of Crude Oil Refinery: Analysis of Metagenomes and Characterization of Isolates. Microorganisms. 2020; 8(5):652. https://doi.org/10.3390/microorganisms8050652
Chicago/Turabian StyleViggor, Signe, Merike Jõesaar, Pedro Soares-Castro, Tanel Ilmjärv, Pedro M. Santos, Atya Kapley, and Maia Kivisaar. 2020. "Microbial Metabolic Potential of Phenol Degradation in Wastewater Treatment Plant of Crude Oil Refinery: Analysis of Metagenomes and Characterization of Isolates" Microorganisms 8, no. 5: 652. https://doi.org/10.3390/microorganisms8050652
APA StyleViggor, S., Jõesaar, M., Soares-Castro, P., Ilmjärv, T., Santos, P. M., Kapley, A., & Kivisaar, M. (2020). Microbial Metabolic Potential of Phenol Degradation in Wastewater Treatment Plant of Crude Oil Refinery: Analysis of Metagenomes and Characterization of Isolates. Microorganisms, 8(5), 652. https://doi.org/10.3390/microorganisms8050652