Deciphering the Role of Colicins during Colonization of the Mammalian Gut by Commensal E. coli

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isogenic Strain Construction
2.2. GFP-Reporter Assays
2.3. Colicin Production Assays
2.4. Colicin Screening Assays
2.5. Animal Experiments
3. Results
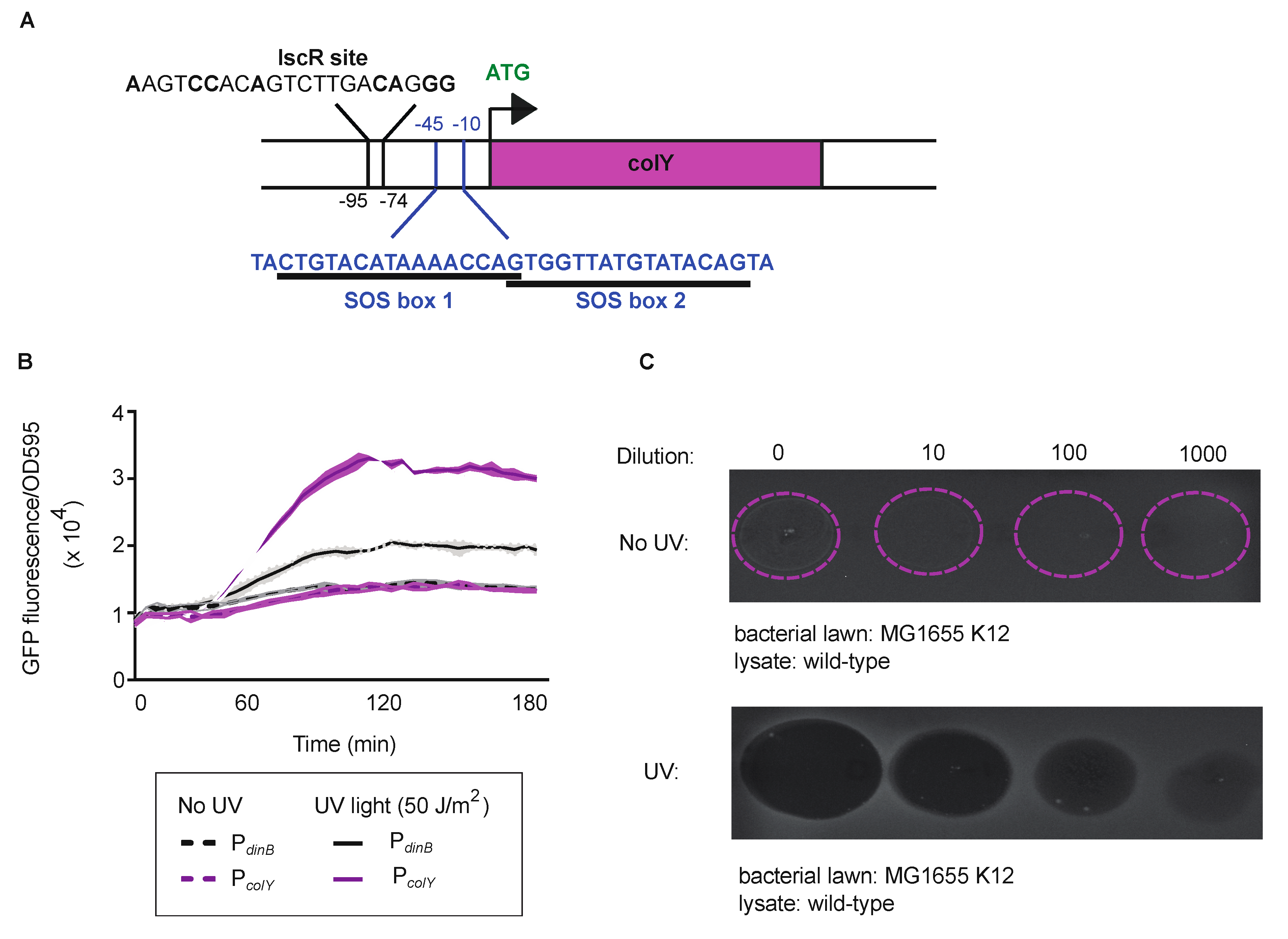
3.1. Regulation and Functionality of the MP1 Colicin
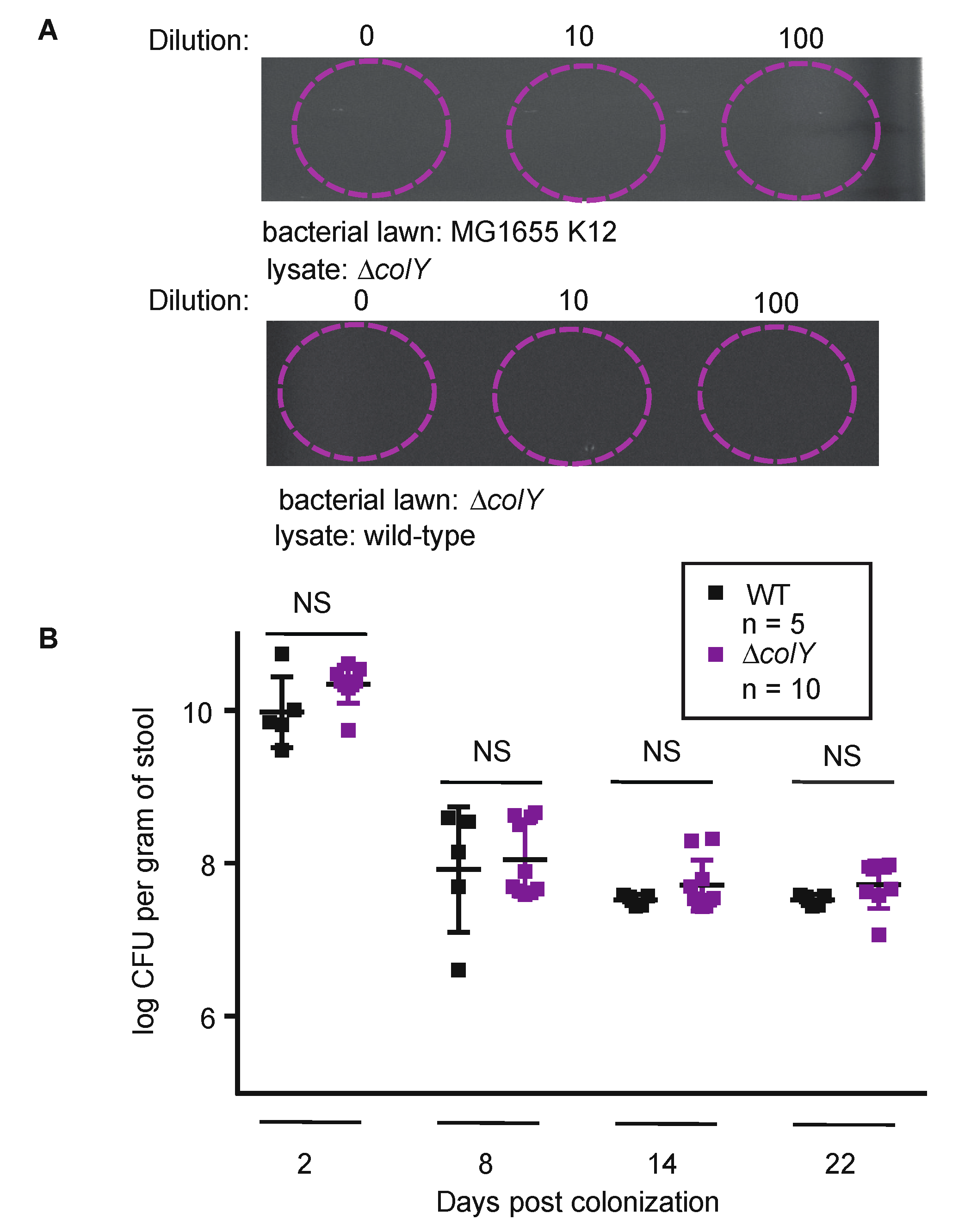
3.2. Sustained Colonization in Healthy Murine Gut Does Not Require Colicin Production
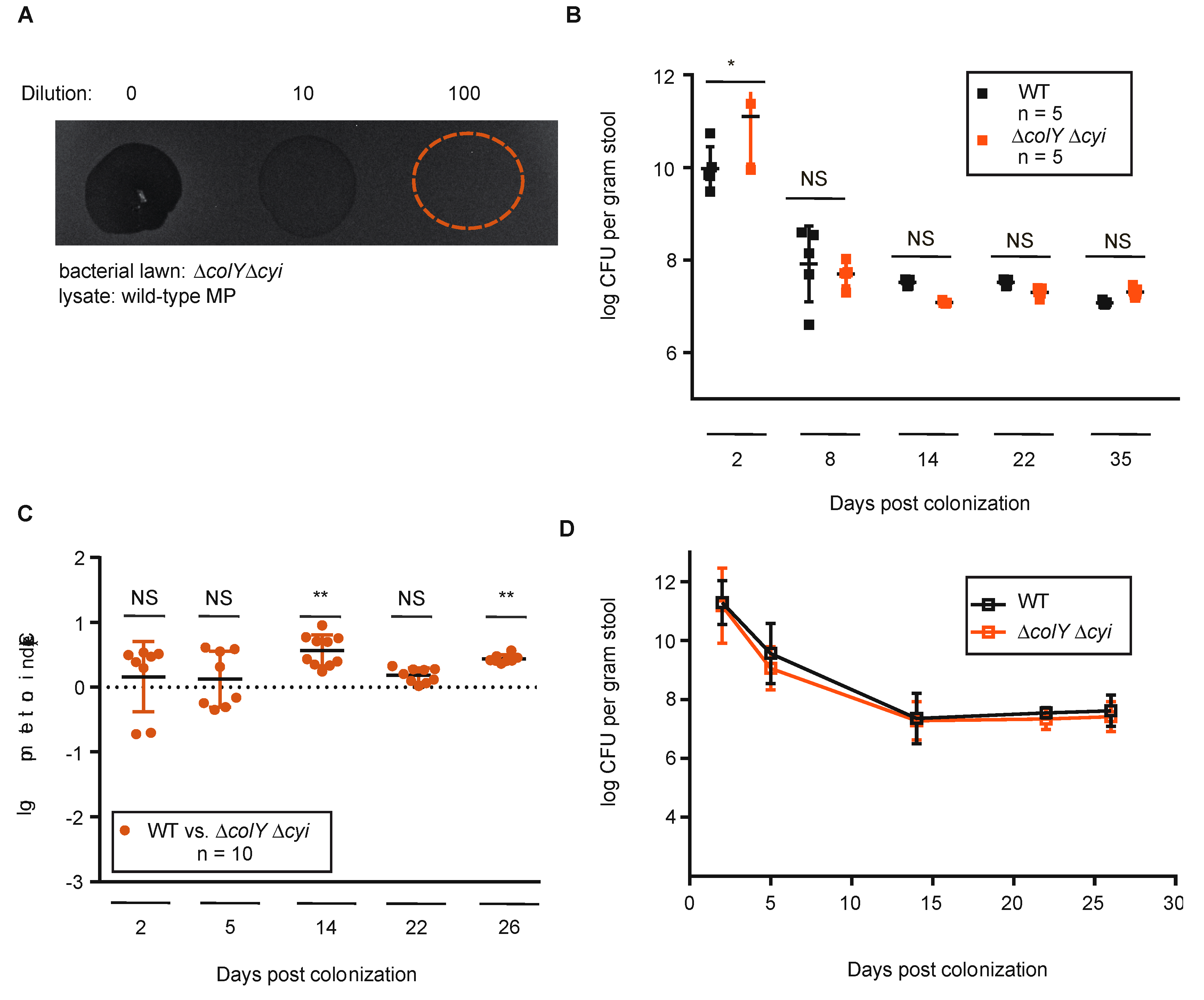
3.3. Sustained Colonization in Healthy Murine Gut Does Not Require Colicin Immunity
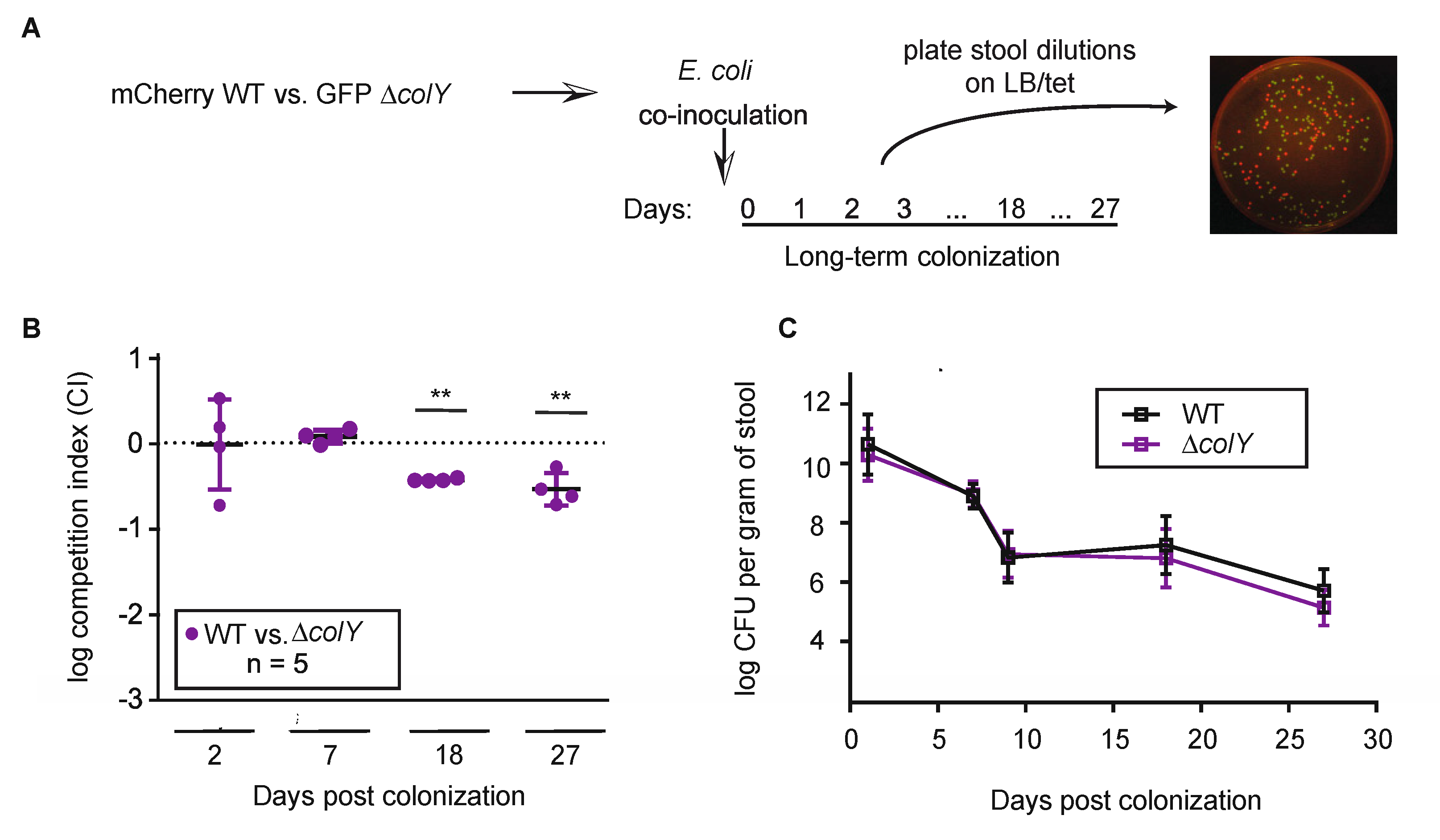
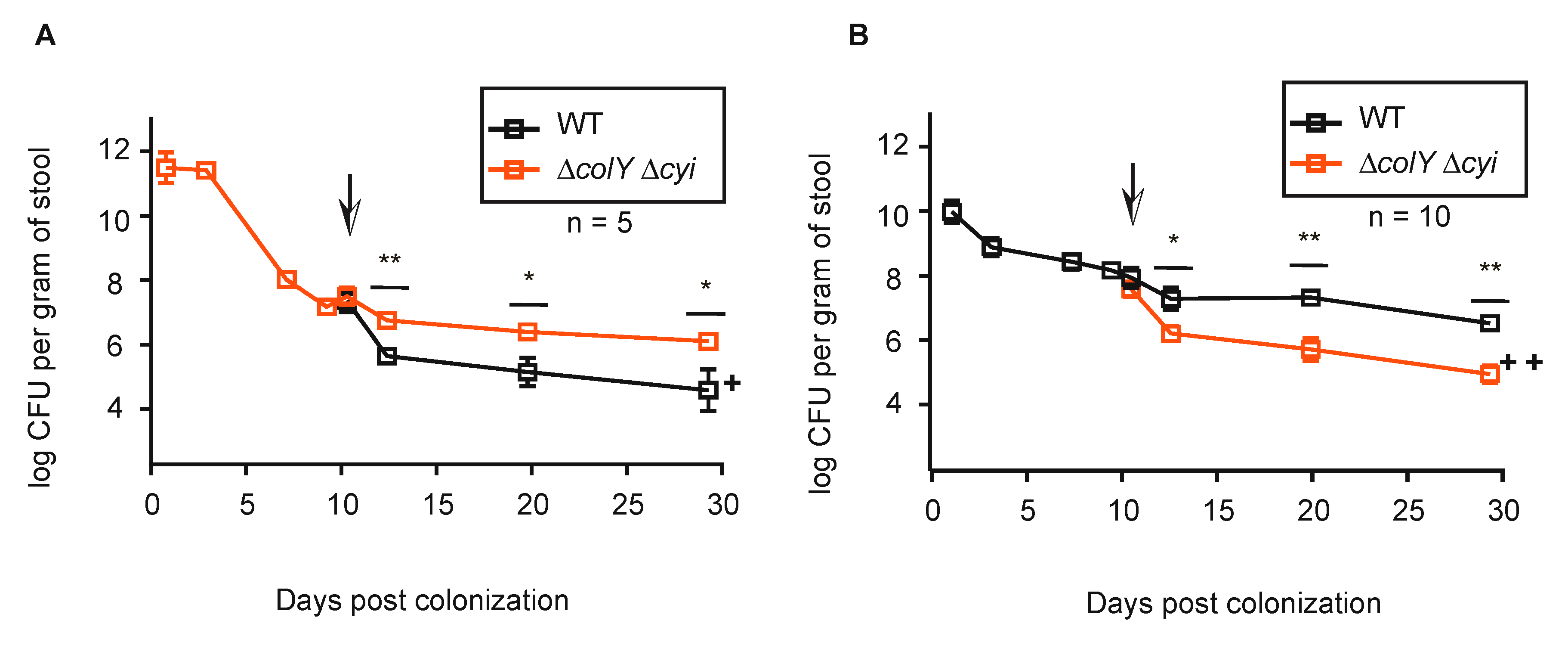
3.4. Withstanding Strain Invasion in the Murine Gut Does Not Require Colicin Production
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kühl, A.A.; Göbel, U.B.; Bereswill, S.; Heimesaat, M.M. Intestinal microbiota shifts towards elevated commensal Escherichia coli loads abrogate colonization resistance against Campylobacter jejuni in mice. PLoS ONE 2012, 7, e35988. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol 2008, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiome shapes intestinal immune respones during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in a microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbendieck, R.M.; Straight, D. Multifaceted interfaces of bacterial competition. J. Bacteriol. 2016, 198, 2145–2155. [Google Scholar] [CrossRef] [Green Version]
- Martens, E.C.; Chiang, H.C.; Gordon, J.I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 2008, 4, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Smalley, D.J.; Tucker, D.L.; Leatham, M.P.; Norris, W.E.; Stevenson, S.J.; Anderson, A.B.; Grissom, J.E.; Laux, D.C.; Cohen, P.S.; et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. USA 2004, 101, 7427–7432. [Google Scholar] [CrossRef] [Green Version]
- Conway, T.; Krogfelt, K.A.; Cohen, P.S. The life of commensal Escherichia coli in the mammalian intestine. EcoSal Plus 2004, 1. [Google Scholar] [CrossRef]
- Wloch-salamon, D.M.; Gerla, D.; Hoekstra, R.F.; de Visser, J.A.G.M. Effect of dispersal and nutrient availability on the competitive ability of toxin-producing yeast. Proc. R Soc. B 2008, 275, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Czaran, T.L.; Hoekstra, R.F.; Paige, L. Chemical warfare between microbes. Proc. Natl. Acad. Sci. USA 2001, 99, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Riley, M.A.; Wertz, J.E. Bacteriocins: Evolution, Ecology, and Application. Annu. Rev. Microbiol. 2002, 56, 117–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascales, E.; Buchanan, S.K.; Duche, D.; Kleanthous, C.; Lloubes, R.; Postle, K.; Riley, M.A.; Slatin, S.; Cavard, D. Colicin Biology. Microbiol. Mol. Biol. Rev. 2002, 71, 158–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillor, O.; Vriezen, J.A.; Riley, M.A. The role of SOS boxes in enteric bacteriocin regulation. Microbiology 2008, 154, 1783–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazaryan, L.; Tonoyan, L.; Ashhab, A.A.; Soares, M.I.M.; Gillor, O. The role of stress in colicin regulation. Arch. Microbiol. 2014, 196, 753–764. [Google Scholar] [CrossRef]
- Jerman, B.; Butala, M.; Zgur-Bertok, D. Sublethal concentrations of ciprofloxacin induce bacteriocin synthesis in Escherichia coli. Antimicrob. Agents Chemother. 2005, 49, 3087–3090. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.; Sabnis, A.; Foster, K.R.; Mavridou, D.A.I. Costs and benefits of provocation in bacterial warfare. Proc. Natl. Acad. Sci. USA 2018, 115, 7593–7598. [Google Scholar] [CrossRef] [Green Version]
- Majeed, H.; Lampert, A.; Ghazaryan, L.; Gillor, O. The weak shall inherit: Bacteriocin-mediated interactions in bacterial populations. PLoS ONE 2013, 8, e63837. [Google Scholar] [CrossRef] [Green Version]
- Kerr, B.; Riley, M.A.; Feldman, M.W.; Bohannan, B.J.M. Local dispersal promotes biodiversity in a real-life game of rock—Paper—Scissors. Nature 2002, 418, 2865–2868. [Google Scholar] [CrossRef]
- Riley, M.A.; Gordon, D.M. A survey of Col plasmids in natural isolates of Escherichia coli and an investigation into the stability of Col-plasmid lineages. J. Gen. Microbiol. 1992, 138, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.M.; Brien, C.L.O. Bacteriocin diversity and the frequency of multiple bacteriocin production in Escherichia coli. Microbiology 2006, 152, 3239–3244. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.M.; Riley, M.A.; Pinou, T. Temporal changes in the frequency of colicinogeny in Escherichia coli from house mice. Microbiology 1998, 144, 2233–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, A.N.; Roggiani, M.; Zhu, J.; Goulian, M.; Kohli, R.M. The SOS response mediates sustained colonization of the mammalian gut. Infect. Immun. 2019, 87, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentges, D.; Pongpech, P.; Que, J. Hypothesis: How streptomycin treatment compromises colonisation resistance against enteric pathogens in mice. Microb. Ecol. Health Dis. 1990, 3, 105–111. [Google Scholar]
- Bazett, M.; Bergeron, M.; Haston, C.K. Streptomycin treatment alters the intestinal microbiome, pulmonary T cell profile and airway hyperresponsiveness in a cystic fibrosis mouse model. Sci. Rep. 2016, 6, 19189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garner, C.D.; Antonopoulos, D.A.; Wagner, B.; Duhamel, G.E.; Keresztes, I.; Ross, D.A.; Young, V.B.; Altier, C. Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica Serovar Typhimurium murine model of infection. Infect. Immun. 2009, 77, 2691–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone-Corsi, M.; Nuccio, S.-P.; Liu, H.; Hernandez, D.; Vu, C.T.; Takahashi, A.A.; Edwards, R.A.; Raffatellu, M. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature 2016, 540, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Myhal, M.; Laux, D.; Cohen, P. Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol. 1982, 1, 186–192. [Google Scholar] [CrossRef]
- Gillor, O.; Giladi, I.; Riley, M.A. Persistence of colicinogenic Escherichia coli in the mouse gastrointestinal tract. BMC Microbiol. 2009, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Kirkup, B.C.; Riley, M.A. Antibiotic-mediated antagonism leads to a bacterial game of rock—Paper—Scissors in vivo. Nature 2004, 428, 694–696. [Google Scholar] [CrossRef]
- Nedialkova, L.P.; Denzler, R.; Koeppel, M.B.; Diehl, M.; Ring, D.; Wille, T.; Gerlach, R.G.; Stecher, B. Inflammation fuels colicin Ib-dependent competition of Salmonella Serovar Typhimurium and E. coli in Enterobacterial blooms. PLoS Pathog. 2014, 10, e1003844. [Google Scholar] [CrossRef] [Green Version]
- Hancock, V.; Dahl, M.; Klemm, P. Probiotic Escherichia coli strain Nissle 1917 outcompetes intestinal pathogens during biofilm formation. J. Medical. Micro. 2010, 59, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasaro, M.; Liu, Z.; Bishar, R.; Kelly, K.; Chattopadhyay, S.; Paul, S.; Sokurenko, E.; Zhu, J.; Goulian, M. Escherichia coli isolate for studying colonization of the mouse intestine and its application to two-component signaling knockouts. J. Bacteriol. 2014, 196, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaslaver, A.; Bren, A.; Ronen, M.; Itzkovitz, S.; Kikoin, I.; Shavit, S.; Liebermeister, W.; Surette, M.G.; Alon, U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 2006, 3, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Culyba, M.J.; Kubiak, J.M.; Mo, C.Y.; Goulian, M.; Kohli, R.M. Non-equilibrium repressor binding kinetics link DNA damage dose to transcriptional timing within the SOS gene network. PLOS Genet. 2018, 14, e100740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, M.A.; Goldstone, C.M.; Wertz, J.E.; Gordon, D. A phylogenetic approach to assessing the targets of microbial warfare. J. Evol. Biol. 2003, 16, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Siryaporn, A.; Goulian, M. Cross-talk suppression between the CpxA—CpxR and EnvZ—OmpR two-component systems in E. coli. Mol. Microbiol. 2008, 70, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Šmajs, D.; Pilsl, H.; Braun, V. Colicin U, a novel colicin produced by Shigella boydii. J. Bacteriol. 1997, 179, 4919–4928. [Google Scholar] [CrossRef] [Green Version]
- Riley, M.A.; Cadavid, L.; Collett, M.S.; Neely, M.N.; Adams, M.D.; Phillips, C.M.; Neel, J.V.; Friedman, D. The newly characterized colicin Y provides evidence of positive selection in pore-former colicin diversification. Microbiology 2000, 146, 1671–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, C.J.; Giel, J.L.; Patschkowski, T.; Luther, C.; Ruzicka, F.J.; Beinert, H.; Kiley, P.J. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 14895–14900. [Google Scholar] [CrossRef] [Green Version]
- Butala, M.; Sonjak, S.; Kamenšek, S.; Hodoscek, M.; Browning, D.F.; Žgur-Bertok, D.; Busby, S.J.W. Double locking of an Escherichia coli promoter by two repressors prevents premature colicin expression and cell lysis. Mol. Med. Rep. 2012, 86, 129–139. [Google Scholar]
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64. [Google Scholar] [PubMed]
- Herschman, H.R.; Donald, A. Comparative study of the events associated with colicin induction. J. Bacteriol. 1967, 94, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Little, J.W. Autodigestion of lexA and phage lambda repressors. Proc. Natl. Acad. Sci. USA 1984, 81, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Mo, C.Y.; Manning, S.A.; Roggiani, M.; Culyba, M.J.; Samuels, A.N.; Sniegowski, P.D.; Goulian, M.; Kohli, R.M. Systematically altering bacterial SOS activity under stress reveals therapeutic strategies for potentiating. mSphere 2016, 1, e00163-16. [Google Scholar] [CrossRef] [Green Version]
- Pennington, J.M.; Rosenberg, S.M. Spontaneous DNA breakage in single living Escherichia coli cells. Nat. Genet. 2007, 39, 797–802. [Google Scholar] [CrossRef] [Green Version]
- Spees, A.M.; Wangdi, T.; Lopez, C.A.; Kingsbury, D.D.; Xavier, M.N.; Winter, S.E.; Tsolis, R.M.; Bäumler, A.J. Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. MBio 2013, 4, e00430-13. [Google Scholar] [CrossRef] [Green Version]
- Antunes, L.C.M.; Han, J.; Ferreira, R.B.R.; Lolic, P.; Borchers, C.H.; Finlay, B.B. Effect of Antibiotic Treatment on the Intestinal Metabolome. Antimicrob. Agents Chemother. 2011, 55, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Shen, T.D.; Chen, E.Z.; Bittinger, K.; Bailey, A.; Roggiani, M.; Sirota-Madi, A.; Friedman, E.S.; Chau, L.; Lin, A.; et al. A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci. Transl. Med. 2017, 9, eaah6888. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Xu, J.; Leip, D.D.; Chen, C.H.; Westover, B.P.; Weatherford, J.; Buhler, J.D.; Gordon, J.I. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 2005, 307, 1955–1959. [Google Scholar] [CrossRef] [Green Version]
- Stecher, B.; Maier, L.; Hardt, W.-D. “Blooming” in the gut: How dysbiosis might contribute to pathogen evolution. Nat. Rev. Microbiol. 2013, 11, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommineni, S.; Bretl, D.J.; Lam, V.; Chakraborty, R.; Simpson, P.; Cao, Y.; Bousounis, P.; Kristich, C.J.; Salzman, N.H. Bacteriocin production augments niche competition by enterococci in the mammalian GI tract. Nature 2015, 526, 719–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, N.; Vardi, S.; Ronen, M.; Alon, U.; Stavans, J. Precise temporal modulation in the response of the SOS DNA repair network in individual bacteria. PLoS Biol. 2005, 3, e238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, B.; Denzler, R.; Maier, L.; Bernet, F.; Sanders, M.J.; Pickard, D.J.; Barthel, M.; Westendorf, A.M.; Krogfelt, K.A.; Walker, A.W.; et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc. Natl. Acad. Sci. USA 2012, 109, 1269–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samuels, A.N.; Roggiani, M.; Smith, K.A.; Zhu, J.; Goulian, M.; Kohli, R.M. Deciphering the Role of Colicins during Colonization of the Mammalian Gut by Commensal E. coli. Microorganisms 2020, 8, 664. https://doi.org/10.3390/microorganisms8050664
Samuels AN, Roggiani M, Smith KA, Zhu J, Goulian M, Kohli RM. Deciphering the Role of Colicins during Colonization of the Mammalian Gut by Commensal E. coli. Microorganisms. 2020; 8(5):664. https://doi.org/10.3390/microorganisms8050664
Chicago/Turabian StyleSamuels, Amanda N., Manuela Roggiani, Kathryn A. Smith, Jun Zhu, Mark Goulian, and Rahul M. Kohli. 2020. "Deciphering the Role of Colicins during Colonization of the Mammalian Gut by Commensal E. coli" Microorganisms 8, no. 5: 664. https://doi.org/10.3390/microorganisms8050664