Revisiting the Metabolic Capabilities of Bifidobacterium longum susbp. longum and Bifidobacterium longum subsp. infantis from a Glycoside Hydrolase Perspective


 ,
,  , and
, and 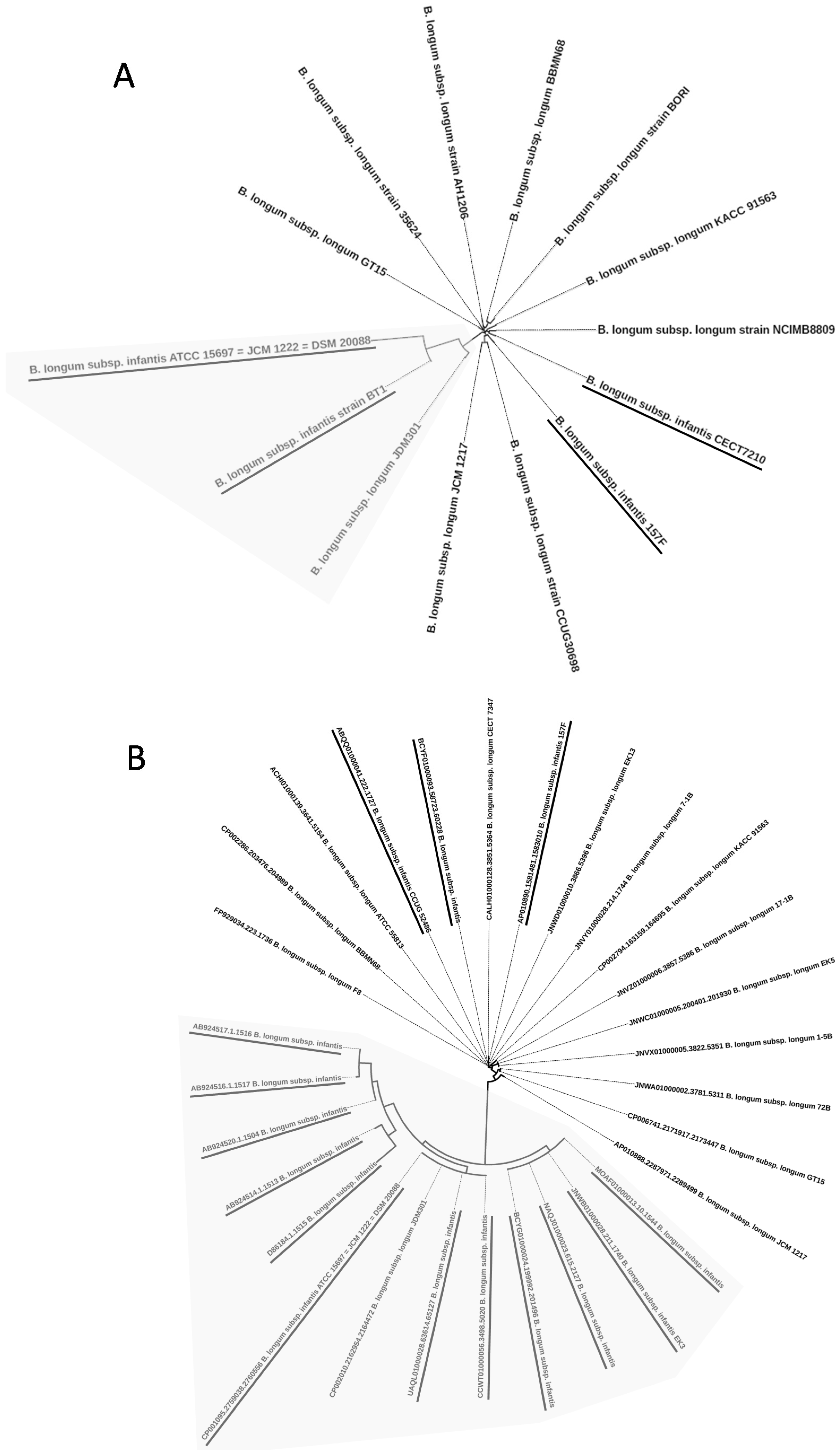{kind=link}
{kind=link}
{kind=link}
{kind=link}
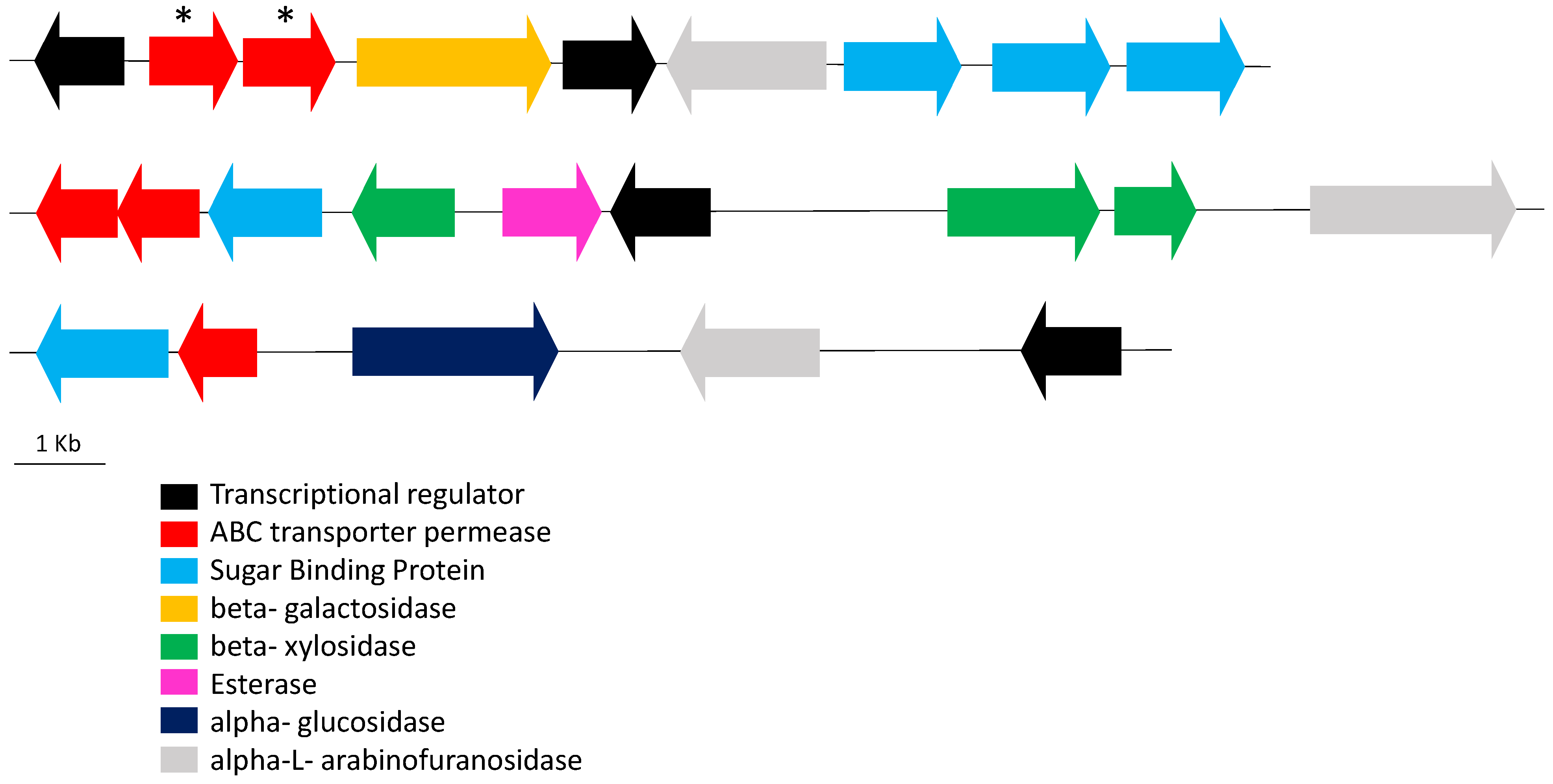{kind=link}
Abstract
:1. Introduction
2. Materials and Methods:
2.1. Phylogenetic Trees Based on 16S rRNA, ldh, recA, and tuf Genes
2.2. Whole Genome Phylogenetic Tree
2.3. Genome Sequences of B. longum
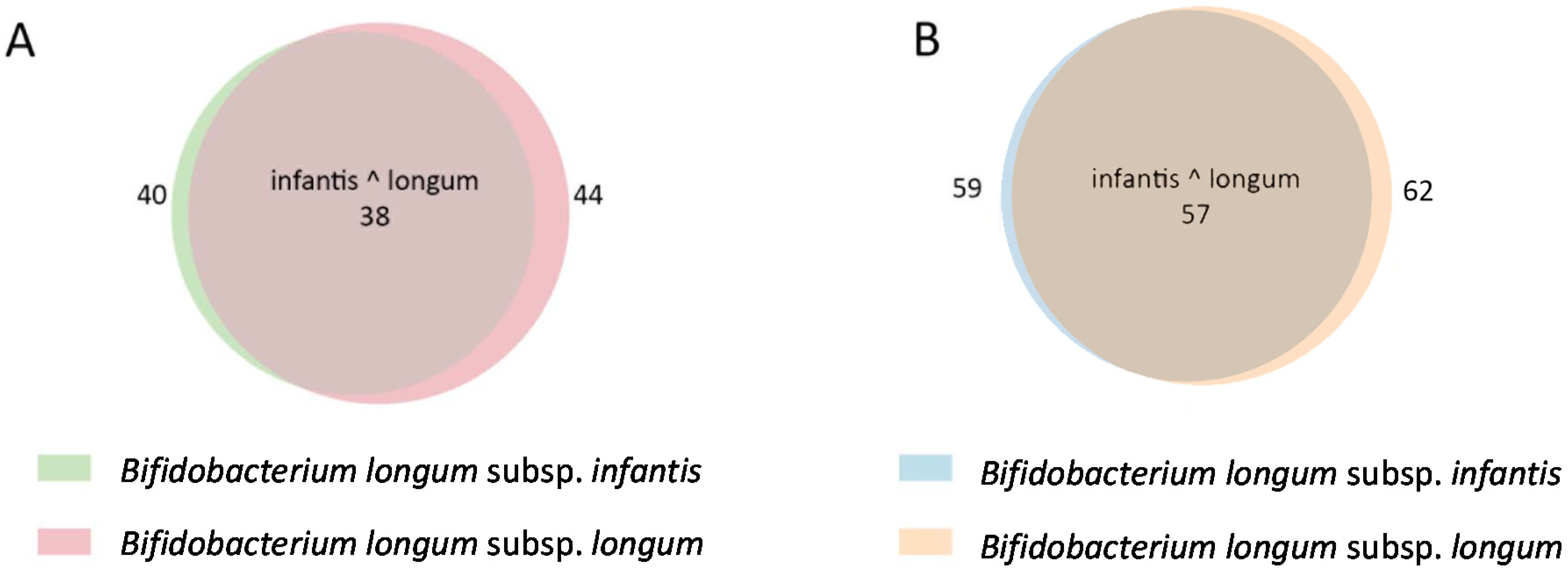
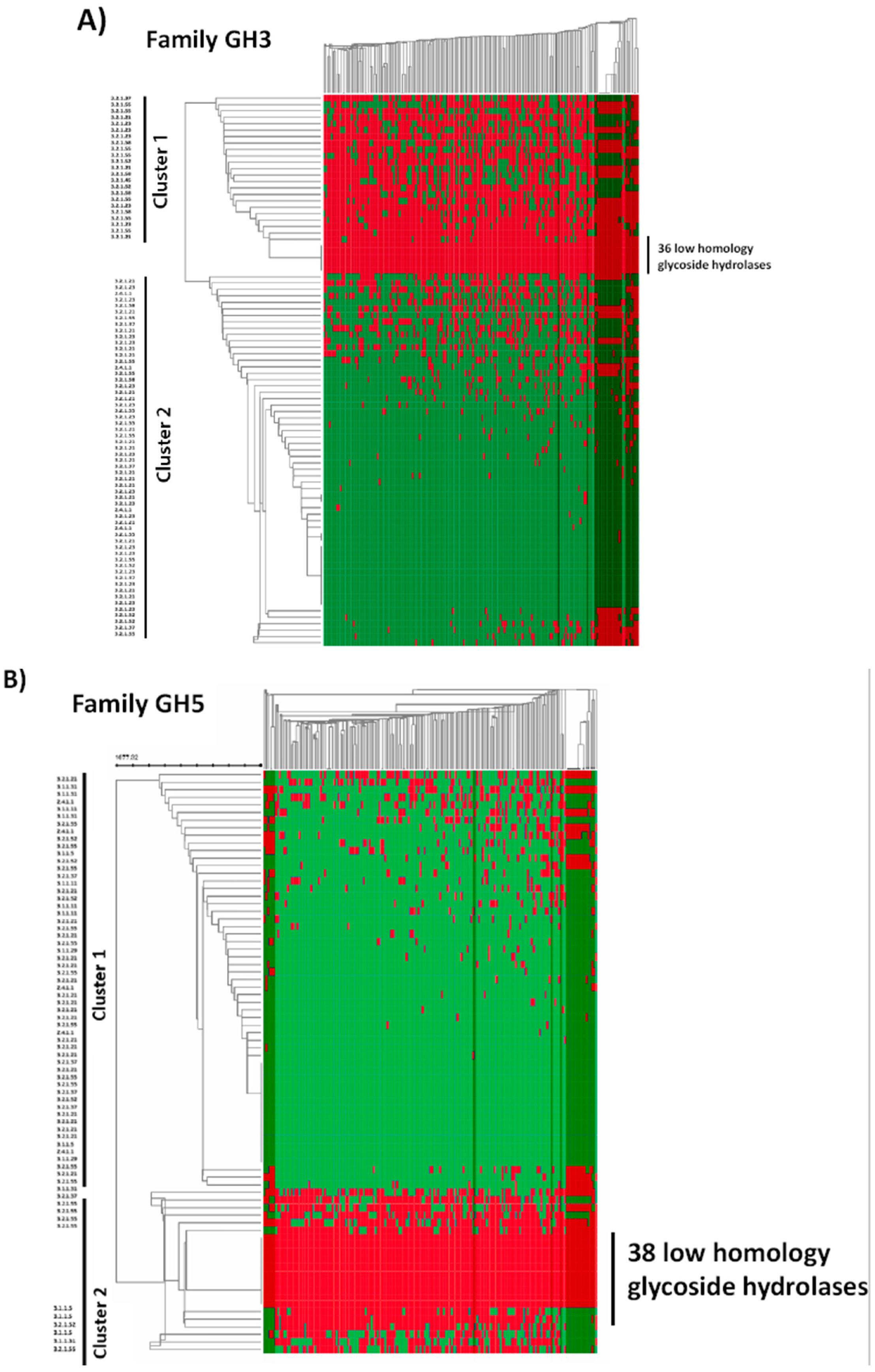
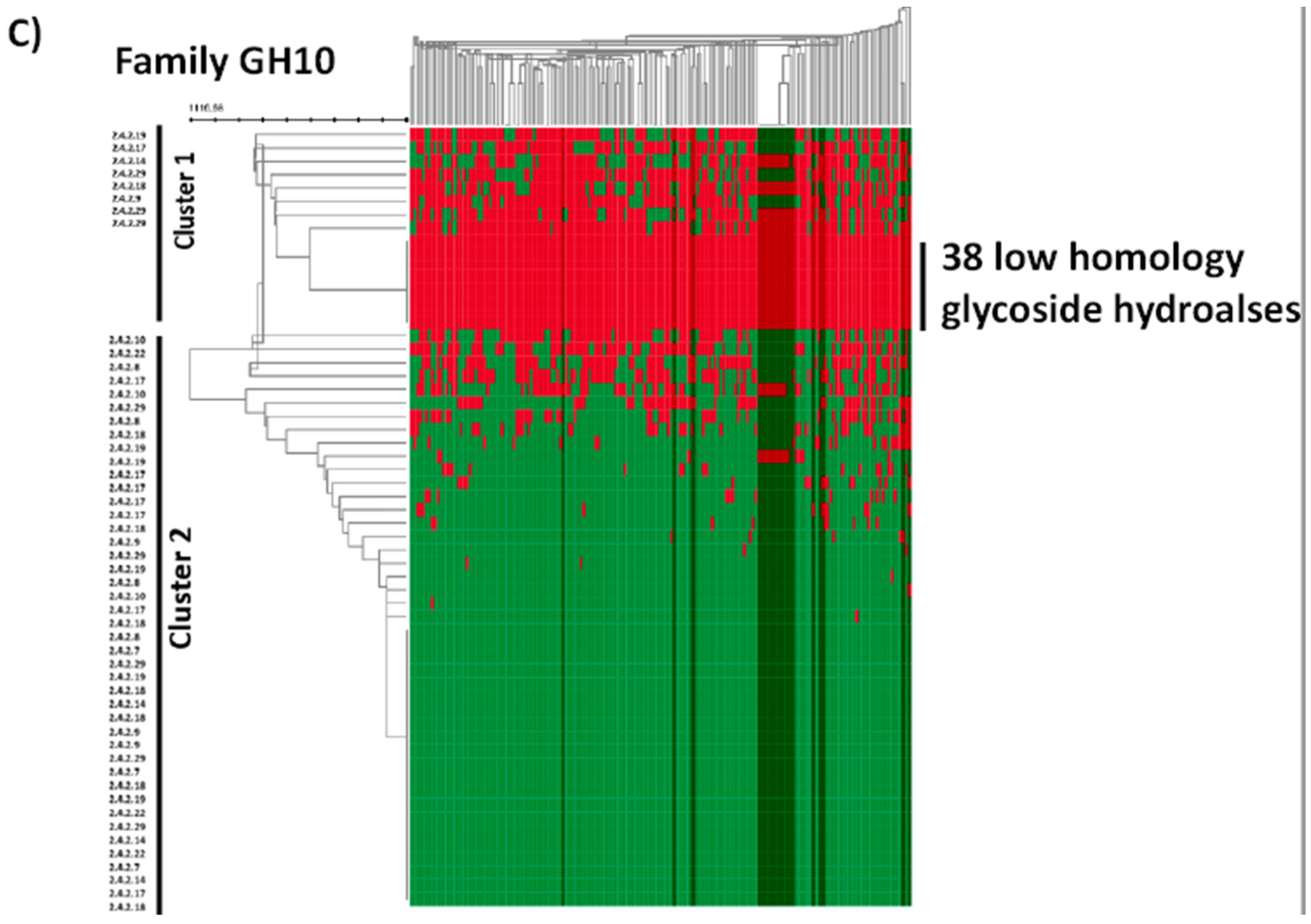
2.4. In Silico Analysis of B. longum GH Capabilities
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bottacini, F.; Ventura, M.; van Sinderen, D.; O’Connell Motherway, M. Diversity, ecology and intestinal function of bifidobacteria. Microb. Cell Fact. 2014, 13 (Suppl. 1), S4. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Koutsoumanis, K.; Allende, A.; Alvarez-Ordoñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F.; Lindqvist, R.; et al. Update of the list of QPS-recommended biological agents intentionally added to food or feed as notified to EFSA. EFSA J. 2019, 15, 5753. [Google Scholar]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Health-Promoting Effects. Microbiol. Spectr. 2018. [Google Scholar] [CrossRef]
- Mattarelli, P.; Bonaparte, C.; Pot, B.; Biavati, B. Proposal to reclassify the three biotypes of Bifidobacterium longum as three subspecies: Bifidobacterium longum subsp. longum subsp. nov., Bifidobacterium longum subsp. infantis comb. nov. and Bifidobacterium longum subsp. suis comb. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Yanokura, E.; Oki, K.; Makino, H.; Modesto, M.; Pot, B.; Mattarelli, P.; Biavati, B.; Watanabe, K. Subspeciation of Bifidobacterium longum by multilocus approaches and amplified fragment length polymorphism: Description of B. longum subsp. suillum subsp. nov.; isolated from the faeces of piglets. Syst. Appl. Microbiol. 2015, 38, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado-Palacio, S.; Arboleya-Montes, S. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turroni, F.; Milani, C.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Viappiani, A.; Lugli, G.A.; Ferrario, C.; Gioiosa, L.; Ferrarini, A.; et al. Deciphering bifidobacterial-mediated metabolic interactions and their impact on gut microbiota by a multi-omics approach. ISME J. 2016, 10, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.A.; Karmirantzou, M.; Snel, B.; Vilanova, D.; Berger, B.; Pessi, G.; Zwahlen, M.C.; Desiere, F.; Bork, P.; Delley, M.; et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2002, 99, 14422–14427. [Google Scholar] [CrossRef] [Green Version]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.H.; Chen, F.; Whitehead, T.R.; Lapidus, A.; Rokhsar, D.S.; Lebrilla, C.B.; German, J.B.; et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964–18969. [Google Scholar] [CrossRef] [Green Version]
- Sela, D.A.; Li, Y.; Lerno, L.; Wu, S.; Marcobal, A.M.; German, J.B.; Chen, X.; Lebrilla, C.B.; Mills, D.A. An infant-associated bacterial commensal utilizes breast milk sialyloligosaccharides. J. Biol. Chem. 2011, 286, 11909–11918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabel, B.; Yde, C.C.; Roos, P.; Marcussen, J.; Jensen, H.M.; Salli, K.; Hirvonen, J.; Ouwehand, A.C.; Morovic, W. Novel Genes and Metabolite Trends in Bifidobacterium longum subsp. infantis Bi-26 Metabolism of Human Milk Oligosaccharide 2’-fucosyllactose. Sci. Rep. 2019, 9, 7983. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, A.; Bottacini, F.; O’Connell Motherway, M.; van Sinderen, D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genom. 2015, 16, 832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, C.; Turroni, F.; Duranti, S.; Lugli, G.A.; Mancabelli, L.; Ferrario, C.; van Sinderen, D.; Ventura, M. Genomics of the Genus Bifidobacterium Reveals Species-Specific Adaptation to the Glycan-Rich Gut Environment. Appl. Environ. Microbiol. 2015, 82, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Blanco, G.; Sánchez, B.; Ruiz, L.; Fdez-Riverola, F.; Margolles, A.; Lourenco, A. Computational approach to the systematic prediction of glycolytic abilities: Looking into human microbiota. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Skuta, C.; Bartůněk, P.; Svozil, D. InCHlib-interactive cluster heatmap for web applications. J. Cheminform. 2014, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Bottacini, F.; Morrissey, R.; Esteban-Torres, M.; James, K.; van Breen, J.; Dikareva, E.; Egan, M.; Lambert, J.; van Limpt, K.; Knol, J.; et al. Comparative genomics and genotype-phenotype associations in Bifidobacterium breve. Sci. Rep. 2018, 8, 10633. [Google Scholar] [CrossRef] [Green Version]
- Kullen, M.J.; Brady, L.J.; O’Sullivan, D.J. Evaluation of using a short region of the recA gene for rapid and sensitive speciation of dominant bifidobacteria in the human large intestine. FEMS Microbiol. Lett. 1997, 154, 377–383. [Google Scholar] [CrossRef]
- Roy, D.; Sirois, S. Molecular differentiation of Bifidobacterium species with amplified ribosomal DNA restriction analysis and alignment of short regions of the ldh gene. FEMS Microbiol. Lett. 2000, 191, 17–24. [Google Scholar] [CrossRef]
- Sakanaka, M.; Nakakawaji, S.; Nakajima, S.; Fukiya, S.; Abe, A.; Saburi, W.; Mori, H.; Yokota, A. A Transposon Mutagenesis System for Bifidobacterium longum subsp. longum Based on an IS3 Family Insertion Sequence, ISBlo11. Appl. Environ. Microbiol. 2018, 84, e00824-18. [Google Scholar] [CrossRef] [Green Version]
- Ventura, M.; Canchaya, C.; Meylan, V.; Klaenhammer, T.R.; Zink, R. Analysis, characterization, and loci of the tuf genes in Lactobacillus and Bifidobacterium species and their direct application for species identification. Appl. Environ. Microbiol. 2003, 69, 6908–6922. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, A.V.; Efimov, B.A.; Smeianov, V.V.; Kafarskaia, L.I.; Pikina, A.P.; Shkoporov, A.N. Intraspecies Genomic Diversity and Long-Term Persistence of Bifidobacterium longum. PLoS ONE 2015, 10, e0135658. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.A.; Chenoll, E.; Casinos, B.; Bataller, E.; Ramón, D.; Genovés, S.; Montava, R.; Ribes, J.M.; Buesa, J.; Fàbrega, J.; et al. Novel probiotic Bifidobacterium longum subsp. infantis CECT 7210 strain active against rotavirus infections. Appl. Environ. Microbiol. 2011, 77, 8775–8783. [Google Scholar]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Mancabelli, L.; Ferrario, C.; Mangifesta, M.; Hevia, A.; Viappiani, A.; Scholz, M.; et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci. Rep. 2015, 5, 5782. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; van Sinderen, D.; Stanton, C. Gene-trait matching across the Bifidobacterium longum pan-genome reveals considerable diversity in carbohydrate catabolism among human infant strains. BMC Genom. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Geirnaert, A.; Van den Abbeele, P.; De Vuyst, L. Complementary Mechanisms for Degradation of Inulin-Type Fructans and Arabinoxylan Oligosaccharides among Bifidobacterial Strains Suggest Bacterial Cooperation. Appl. Environ. Microbiol. 2018, 84, e02893-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastell, H.; Westermann, P.; Meyer, A.S.; Tuomainen, P.; Tenkanen, M. In vitro fermentation of arabinoxylan-derived carbohydrates by bifidobacteria and mixed fecal microbiota. J. Agric. Food Chem. 2009, 57, 8598–8606. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aceituno, L.; Esteban-Torres, M.; James, K.; Moreno, F.J.; van Sinderen, D. Metabolism of biosynthetic oligosaccharides by human-derived Bifidobacterium breve UCC2003 and Bifidobacterium longum NCIMB 8809. Int. J. Food Microbiol. 2020, 316, 108476. [Google Scholar] [CrossRef]
- Garrido, D.; Ruiz-Moyano, S.; Kirmiz, N.; Davis, J.C.; Totten, S.M.; Lemay, D.G.; Ugalde, J.A.; German, J.B.; Lebrilla, C.B.; Mills, D.A. A novel gene cluster allows preferential utilization of fucosylated milk oligosaccharides in Bifidobacterium longum subsp. longum SC596. Sci. Rep. 2016, 6, 35045. [Google Scholar] [CrossRef] [Green Version]
- Sakanaka, M.; Hansen, M.E.; Gotoh, A.; Katoh, T.; Yoshida, K.; Odamaki, T.; Yachi, H.; Sugiyama, Y.; Kurihara, S.; Hirose, J.; et al. Evolutionary adaptation in fucosyllactose uptake systems supports bifidobacteria-infant symbiosis. Sci. Adv. 2019, 5, eaaw7696. [Google Scholar] [CrossRef] [Green Version]
- Margolles, A.; de los Reyes-Gavilán, C.G. Purification and functional characterization of a novel alpha-l-arabinofuranosidase from Bifidobacterium longum B667. Appl. Environ. Microbiol. 2003, 69, 5096–5103. [Google Scholar] [CrossRef] [Green Version]
- Komeno, M.; Hayamizu, H.; Fujita, K.; Ashida, H. Two Novel α-l-Arabinofuranosidases from Bifidobacterium longum subsp. longum Belonging to Glycoside Hydrolase Family 43 Cooperatively Degrade Arabinan. Appl. Environ. Microbiol. 2019, 85, e02582-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K.; Sasaki, Y.; Kitahara, K. Degradation of plant arabinogalactan proteins by intestinal bacteria: Characteristics and functions of the enzymes involved. Appl. Microbiol. Biotechnol. 2019, 103, 7451–7457. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, K.M.; Beldman, G.; Voragen, A.G. A new arabinofuranohydrolase from Bifidobacterium adolescentis able to remove arabinosyl residues from double-substituted xylose units in arabinoxylan. Appl. Microbiol. Biotechnol. 1997, 47, 231–235. [Google Scholar] [CrossRef]
- Broekaert, W.F.; Courtin, C.M.; Verbeke, K.; Van de Wiele, T.; Verstraete, W.; Delcour, J.A. Prebiotic and other health-related effects of cereal-derived arabinoxylans, arabinoxylan-oligosaccharides, and xylooligosaccharides. Crit. Rev. Food Sci. Nutr. 2011, 51, 178–194. [Google Scholar] [CrossRef]
- Walton, G.E.; Lu, C.; Trogh, I.; Arnaut, F.; Gibson, G.R. A randomised, double-blind, placebo controlled cross-over study to determine the gastrointestinal effects of consumption of arabinoxylan-oligosaccharides enriched bread in healthy volunteers. Nutr. J. 2012, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Rivière, A.; Gagnon, M.; Weckx, S.; Roy, D.; De Vuyst, L. Mutual Cross-Feeding Interactions between Bifidobacterium longum subsp. longum NCC2705 and Eubacterium rectale ATCC 33656 Explain the Bifidogenic and Butyrogenic Effects of Arabinoxylan Oligosaccharides. Appl. Environ. Microbiol. 2015, 81, 7767–7781. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, G.; Ruiz, L.; Tamés, H.; Ruas-Madiedo, P.; Fdez-Riverola, F.; Sánchez, B.; Lourenço, A.; Margolles, A. Revisiting the Metabolic Capabilities of Bifidobacterium longum susbp. longum and Bifidobacterium longum subsp. infantis from a Glycoside Hydrolase Perspective. Microorganisms 2020, 8, 723. https://doi.org/10.3390/microorganisms8050723
Blanco G, Ruiz L, Tamés H, Ruas-Madiedo P, Fdez-Riverola F, Sánchez B, Lourenço A, Margolles A. Revisiting the Metabolic Capabilities of Bifidobacterium longum susbp. longum and Bifidobacterium longum subsp. infantis from a Glycoside Hydrolase Perspective. Microorganisms. 2020; 8(5):723. https://doi.org/10.3390/microorganisms8050723
Chicago/Turabian StyleBlanco, Guillermo, Lorena Ruiz, Hector Tamés, Patricia Ruas-Madiedo, Florentino Fdez-Riverola, Borja Sánchez, Anália Lourenço, and Abelardo Margolles. 2020. "Revisiting the Metabolic Capabilities of Bifidobacterium longum susbp. longum and Bifidobacterium longum subsp. infantis from a Glycoside Hydrolase Perspective" Microorganisms 8, no. 5: 723. https://doi.org/10.3390/microorganisms8050723
APA StyleBlanco, G., Ruiz, L., Tamés, H., Ruas-Madiedo, P., Fdez-Riverola, F., Sánchez, B., Lourenço, A., & Margolles, A. (2020). Revisiting the Metabolic Capabilities of Bifidobacterium longum susbp. longum and Bifidobacterium longum subsp. infantis from a Glycoside Hydrolase Perspective. Microorganisms, 8(5), 723. https://doi.org/10.3390/microorganisms8050723