Efficient Genome Editing in Bacillus licheniformis Mediated by a Conditional CRISPR/Cas9 System

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Reagents
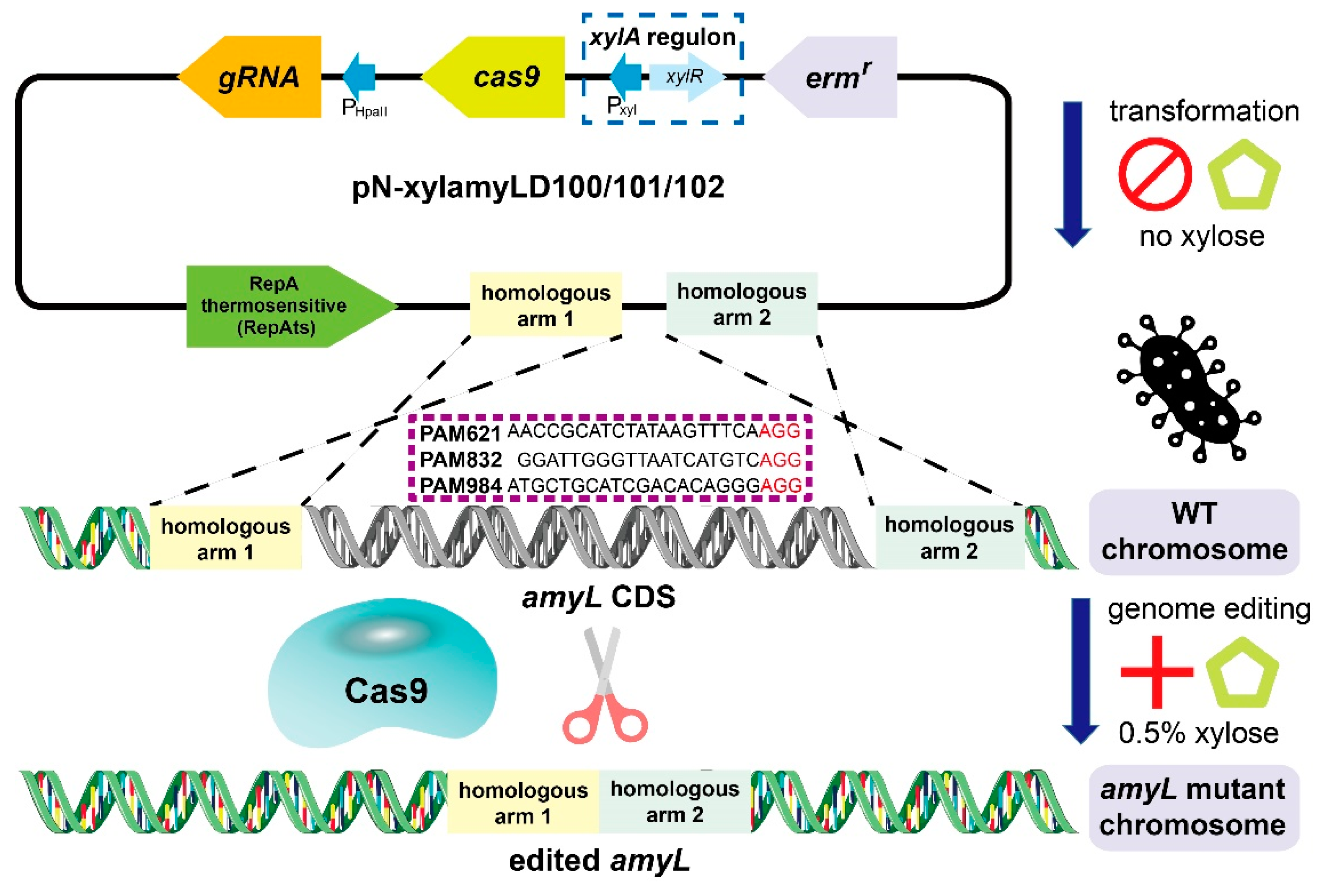
2.3. Recombinant Vector Construction
2.4. Transformation of the All-in-One Conditional CRISPR/Cas9 Plasmid and Conditional Genome Editing
2.5. Biomass and Amylase Assay
2.6. Statistical Analysis
3. Results
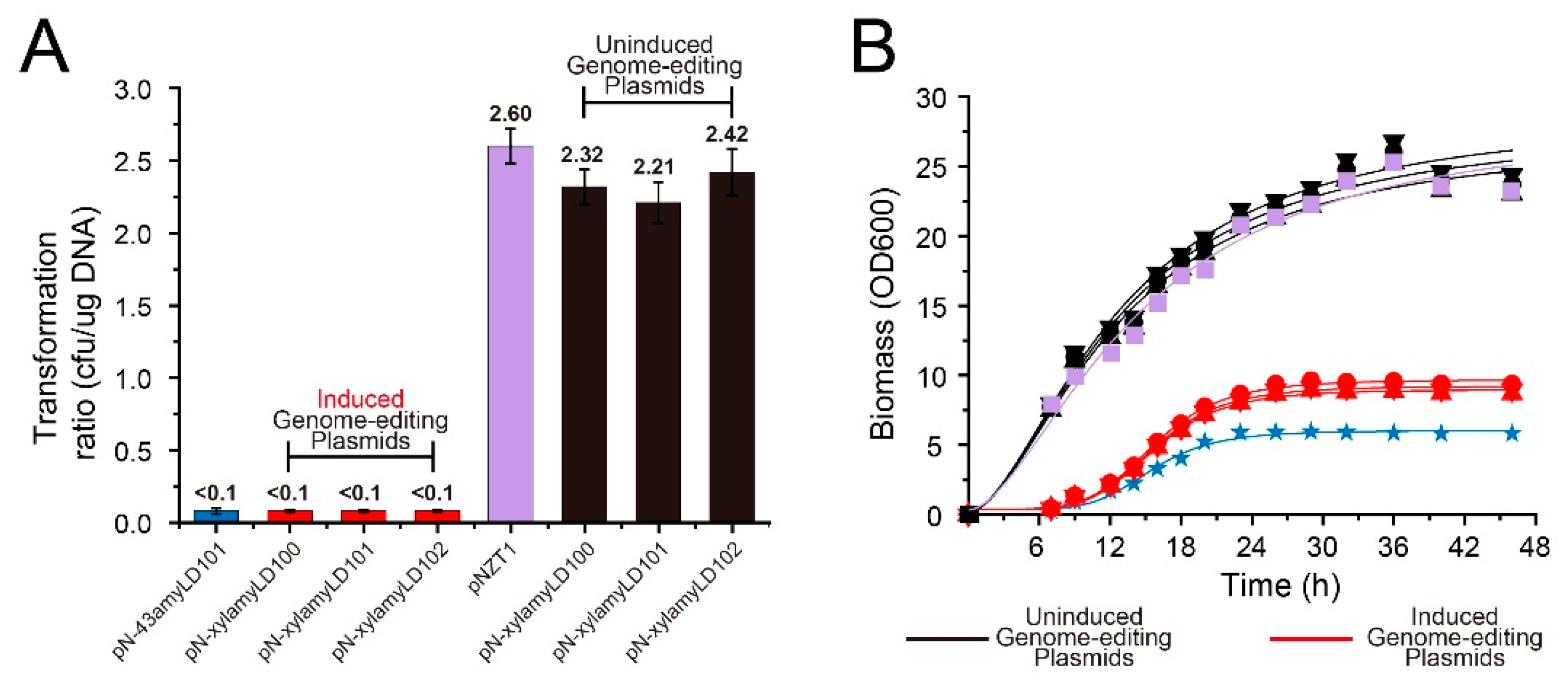
3.1. Inactivation of cas9 Transcription Contributes to the High Transformation Rates of the Genome-Editing Plasmids
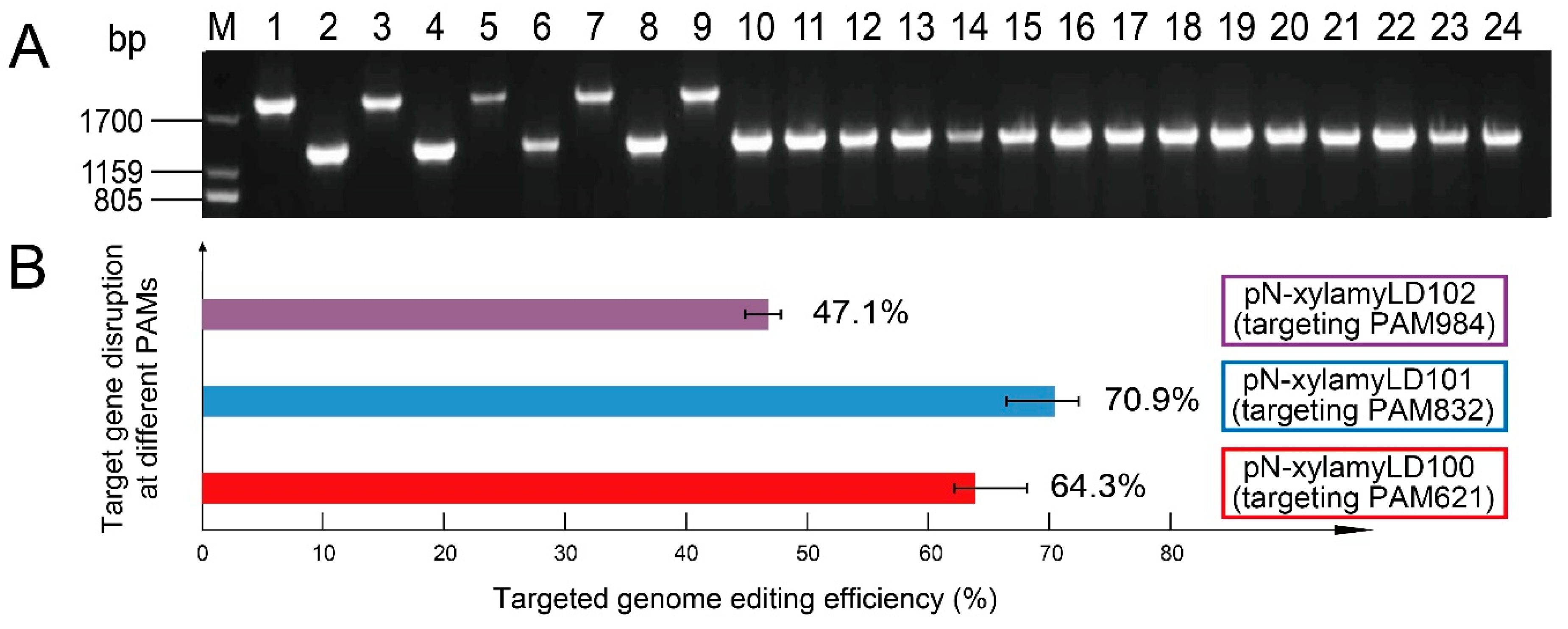
3.2. Efficient Genome Editing Was Triggered by 0.5% Xylose
3.3. Low Temperatures Increased the Genome-Editing Efficiency
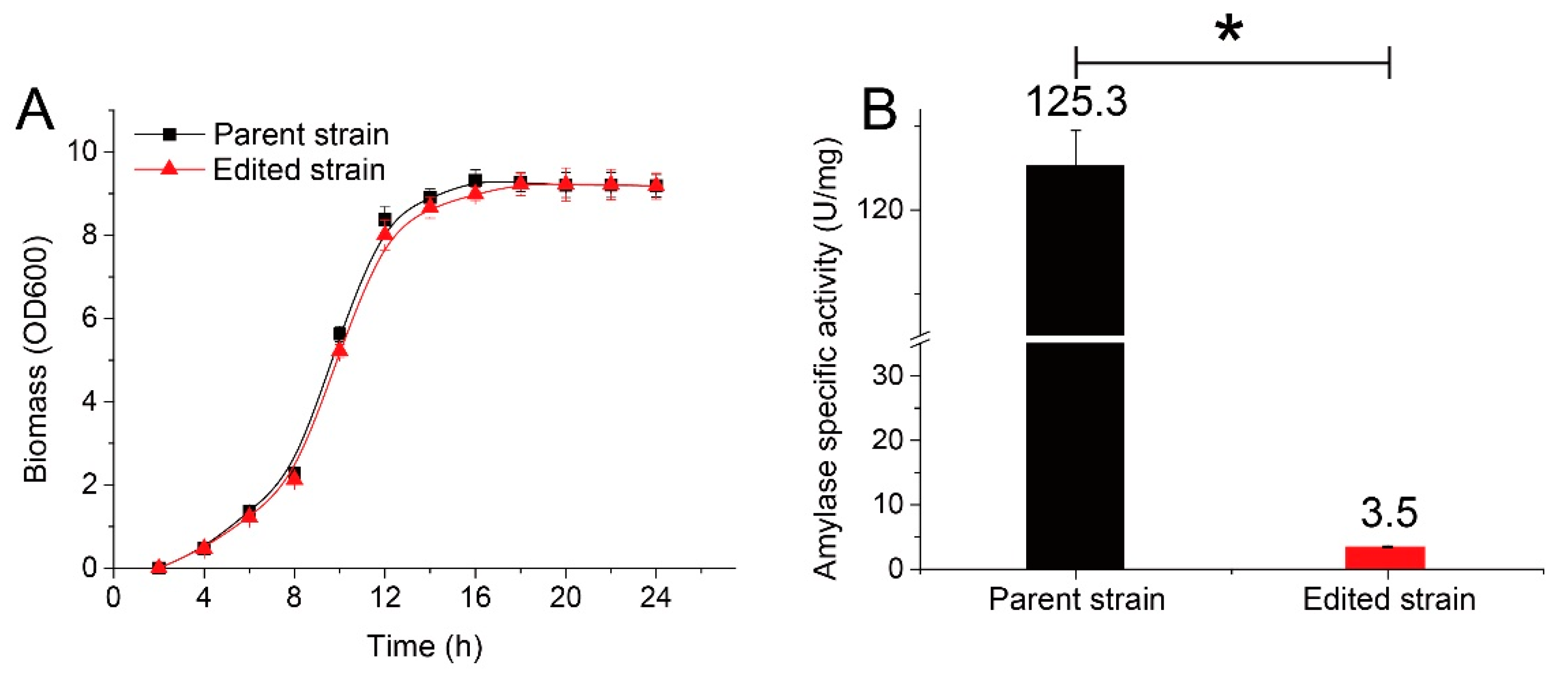
3.4. The Genome-Edited Strain Exhibited the Predicted Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, Y.; Jin, K.; Zhang, L.; Ding, Z.; Gu, Z.; Shi, G. Development of an Inducible Secretory Expression System in Bacillus licheniformis Based on an Engineered Xylose Operon. J. Agric. Food Chem. 2018, 66, 9456–9464. [Google Scholar] [CrossRef]
- Rey, M.W.; Ramaiya, P.; Nelson, B.A.; Brody-Karpin, S.D.; Zaretsky, E.J.; Tang, M.; De Leon, A.L.; Xiang, H.; Gusti, V.; Clausen, I.G.; et al. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Boil. 2004, 5, r77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Liu, H.; Yuan, F.; Chai, H.; Wang, H.; Liu, F.; Li, Y.; Zhang, H.; Lu, F. Development and application of a CRISPR/Cas9 system for Bacillus licheniformis genome editing. Int. J. Boil. Macromol. 2019, 122, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Degering, C.; Eggert, T.; Puls, M.; Bongaerts, J.; Evers, S.; Maurer, K.-H.; Jaeger, K.-E. Optimization of Protease Secretion in Bacillus subtilis and Bacillus licheniformis by Screening of Homologous and Heterologous Signal Peptides. Appl. Environ. Microbiol. 2010, 76, 6370–6376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijland, R.; Burgess, J.G.; Errington, J.; Veening, J.-W. Transformation of Environmental Bacillus subtilis Isolates by Transiently Inducing Genetic Competence. PLoS ONE 2010, 5, e9724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, K.; Wollherr, A.; Larsen, M.; Rachinger, M.; Liesegang, H.; Ehrenreich, A.; Meinhardt, F. Facilitation of Direct Conditional Knockout of Essential Genes in Bacillus licheniformis DSM13 by Comparative Genetic Analysis and Manipulation of Genetic Competence. Appl. Environ. Microbiol. 2010, 76, 5046–5057. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, A.; Boudet, J.; Berzigotti, S.; Duval, V.; Teller, N.; Mengin-Lecreulx, D.; Luxen, A.; Simorre, J.-P.; Joris, B. A Peptidoglycan Fragment Triggers β-lactam Resistance in Bacillus licheniformis. PLoS Pathog. 2012, 8, e1002571. [Google Scholar] [CrossRef] [Green Version]
- Voigt, B.; Albrecht, D.; Sievers, S.; Becher, D.; Bongaerts, J.; Evers, S.; Schweder, T.; Maurer, K.-H.; Hecker, M. High-resolution proteome maps of Bacillus licheniformis cells growing in minimal medium. Proteomics 2015, 15, 2629–2633. [Google Scholar] [CrossRef]
- Kostner, D.; Rachinger, M.; Liebl, W.; Ehrenreich, A. Markerless deletion of putative alanine dehydrogenase genes in Bacillus licheniformis using a codBA-based counterselection technique. Microbiology 2017, 163, 1532–1539. [Google Scholar] [CrossRef]
- Li, K.; Cai, D.; Wang, Z.; He, Z.; Chen, S. Development of an Efficient Genome Editing Tool in Bacillus licheniformis Using CRISPR-Cas9 Nickase. Appl. Environ. Microbiol. 2018, 84, e02608-17. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.E.; Dupuis, M.-È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Spilman, M.; Cocozaki, A.; Hale, C.; Shao, Y.; Ramia, N.; Terns, R.; Terns, M.; Li, H.; Stagg, S. Structure of an RNA Silencing Complex of the CRISPR-Cas immune system. Mol. Cell 2013, 52, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrascia, M.; D’Addabbo, P.; Roberto, R.; Porcelli, F.; Oliva, M.; Calia, C.; Dionisi, A.M.; Pazzani, C. Characterization of CRISPR-Cas Systems in Serratia marcescens Isolated from Rhynchophorus ferrugineus (Olivier, 1790) (Coleoptera: Curculionidae). Microorganisms 2019, 7, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Boil. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Westbrook, A.W.; Moo-Young, M.; Chou, C.P. Development of a CRISPR-Cas9 Tool Kit for Comprehensive Engineering of Bacillus subtilis. Appl. Environ. Microbiol. 2016, 82, 4876–4895. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.-J.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR–Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef]
- Drejer, E.B.; Hakvåg, S.; Irla, M.; Brautaset, T. Genetic Tools and Techniques for Recombinant Expression in Thermophilic Bacillaceae. Microorganisms 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Hornbak, T. The effect of inoculum age and solid versus liquid propagation on inoculum quality of an industrial Bacillus licheniformis strain. FEMS Microbiol. Lett. 2004, 236, 145–151. [Google Scholar] [CrossRef]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef] [PubMed]
- Zakataeva, N.P.; Nikitina, O.V.; Gronskiy, S.V.; Romanenkov, D.V.; Livshits, V.A. A simple method to introduce marker-free genetic modifications into the chromosome of naturally nontransformable Bacillus amyloliquefaciens strains. Appl. Microbiol. Biotechnol. 2009, 85, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Xu, H.; Xie, L.; He, M.; Hou, H.; Wu, C.; Li, Y.; Zhang, C. Effects of Nitrogen Level during Seed Production on Wheat Seed Vigor and Seedling Establishment at the Transcriptome Level. Int. J. Mol. Sci. 2018, 19, 3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczelkun, M.D.; Tikhomirova, M.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Liu, J.-J.; Osuna, B.A.; Xu, M.; Berry, J.D.; Rauch, B.; Nogales, E.; Bondy-Denomy, J.; Doudna, J.A. Temperature-Responsive Competitive Inhibition of CRISPR-Cas9. Mol. Cell 2019, 73, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Xiang, G.; Zhang, X.; An, C.; Cheng, C.; Wang, H. Temperature effect on CRISPR-Cas9 mediated genome editing. J. Genet. Genom. 2017, 44, 199–205. [Google Scholar] [CrossRef]
- Høyland-Kroghsbo, N.M.; Muñoz, K.A.; Bassler, B.L.; Gottesman, S.; Marraffini, L. Temperature, by Controlling Growth Rate, Regulates CRISPR-Cas Activity in Pseudomonas aeruginosa. mBio 2018, 9, e02184-18. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.; Aishan, T.; Maruyama, J.-I.; Kitamoto, K. Enhanced Production and Secretion of Heterologous Proteins by the Filamentous Fungus Aspergillus oryzae via Disruption of Vacuolar Protein Sorting Receptor Gene Aovps10. Appl. Environ. Microbiol. 2010, 76, 5718–5727. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-B.; Timmusk, S. A Simplified Method for Gene Knockout and Direct Screening of Recombinant Clones for Application in Paenibacillus polymyxa. PLoS ONE 2013, 8, e68092. [Google Scholar] [CrossRef] [Green Version]
- Kabisch, J.; Pratzka, I.; Meyer, H.; Albrecht, D.; Lalk, M.; Ehrenreich, A.; Schweder, T. Metabolic engineering of Bacillus subtilis for growth on overflow metabolites. Microb. Cell Factories 2013, 12, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.C.; Cha, J.H.; Kim, J.R.; Jang, S.Y.; Seo, B.C.; Cheong, T.K.; Lee, D.S.; Choi, Y.D.; Park, K.H. Catalytic properties of the cloned amylase from Bacillus licheniformis. J. Boil. Chem. 1992, 267, 22108–22114. [Google Scholar]
- Cha, H.-J.; Yoon, H.-G.; Kim, Y.-W.; Lee, H.-S.; Kim, J.-W.; Kweon, K.-S.; Oh, B.-H.; Park, K.-H. Molecular and enzymatic characterization of a maltogenic amylase that hydrolyzes and transglycosylates acarbose. JBIC J. Boil. Inorg. Chem. 1998, 253, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef]
- Wu, W.Y.; Lebbink, J.H.G.; Kanaar, R.; Geijsen, N.; Van Der Oost, J. Genome editing by natural and engineered CRISPR-associated nucleases. Nat. Methods 2018, 14, 642–651. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Manna, D.; Maji, B.; Gangopadhyay, S.A.; Cox, K.J.; Zhou, Q.; Law, B.K.; Mazitschek, R.; Choudhary, A. A Singular System with Precise Dosing and Spatiotemporal Control of CRISPR-Cas9. Angew. Chem. Int. Ed. 2019, 58, 6285–6289. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-based Photoactivatable Transcription System. Chem. Boil. 2015, 22, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.; Dadon, D.B.; Cheng, A.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.; Mogk, A.; Schumann, W. A xylose-inducible Bacillus subtilis integration vector and its application. Gene 1996, 181, 71–76. [Google Scholar] [CrossRef]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.-H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.V.; Sternberg, S.H.; Taylor, D.W.; Staahl, B.T.; Bardales, J.A.; Kornfeld, J.E.; Doudna, J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 2015, 112, 2984–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, X.; Zhang, L.; Ding, Z.; Xu, S.; Gu, Z.; Shi, G. Transcriptional Changes in the Xylose Operon in Bacillus licheniformis and Their Use in Fermentation Optimization. Int. J. Mol. Sci. 2019, 20, 4615. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or Plasmid | Description 1 | Source/Reference 2, 3 |
|---|---|---|
| Strains | ||
| Escherichia coli JM109 | F′, traD36, proAB +. lacIq, Δ (lacZ), M15/Δ (lac-proAB), glnV44, e14−, gyrA96, recA1, relA1, endA1, thi, hsdR17 | CICIM-CU |
| Bacillus licheniformis DSM13 | Wild-type | CICIM-CU |
| BLG100 | B. licheniformis DSM13 harboring pNZT1 | This work |
| BLG107 | B. licheniformis DSM13 harboring pN-xylamyLD100 | This work |
| BLG108 | B. licheniformis DSM13 harboring pN-xylamyLD101 | This work |
| BLG109 | B. licheniformis DSM13 harboring pN-xylamyLD102 | This work |
| BLG110 | B. licheniformis DSM13 harboring pN-43amyLD100 | This work |
| Plasmids | ||
| pMD19-T | E. coli cloning vector, ApR, 2692 bp | TaKaRa |
| pCas9 | E. coli vector with a minimal CRISPR3-Cas9, the leader sequence, and a repeat-spacer region, 12671 bp | [21] |
| pE194 | Replication-thermosensitive Bacillus vector, 3728 bp | [22] |
| pNZT1 | pE194-derivative, Bacillus vector, EmR, 4069 bp | [22] |
| pMA5 | E. coli/Bacillus shuttle vector, NeoR/ApR, PHpaII, 7107 bp | BGSC |
| pHY300-PLK | E. coli/Bacillus shuttle vector, ApR/TetR, 4870 bp | CICIM-CU |
| pN-sgRNA1 | pNZT1 derivative with an sgRNA targeting the PAM621 locus, 4550 bp | This work |
| pN-sgRNA2 | pNZT1 derivative with an sgRNA targeting the PAM832 locus, 4549 bp | This work |
| pN-sgRNA3 | pNZT1 derivative with an sgRNA targeting the PAM984 locus, 4549 bp | This work |
| pM-xylACas9 | pMD18 derivative with the xylose regulon and cas9, ApR, 8232 bp | This work |
| pN-sgRNA1-xylACas9 | pNZT1 derivative with an sgRNA targeting the PAM621 locus and cas9 under the control of the xylose regulon, 10090 bp | This work |
| pN-sgRNA2-xylACas9 | pNZT1 derivative with an sgRNA targeting the PAM832 locus and cas9 under the control of the xylose regulon, 10089 bp | This work |
| pN-sgRNA3-xylACas9 | pNZT1 derivative with an sgRNA targeting the PAM984 locus and cas9 under the control of the xylose regulon, 10089 bp | This work |
| pN-xylamyLD100 | pNZT1-derivative genome-editing plasmid targeting the PAM621 locus of amyl with cas9 under the control of the xylose regulon, 10880 bp | This work |
| pN-xylamyLD101 | pNZT1-derivative genome-editing plasmid targeting the PAM832 locus of amyl with cas9 under the control of the xylose regulon, 10879 bp | This work |
| pN-xylamyLD102 | pNZT1-derivative genome-editing plasmid targeting the PAM984 locus of amyl with cas9 under the control of the xylose regulon, 10879 bp | This work |
| pN-43amyLD100 | pNZT1-derivative genome-editing plasmid targeting the PAM621 locus of amyl with cas9 under the control of the P43 promoter, 9758 bp | Our Lab |
| Primer | Sequence (5′-3′) |
|---|---|
| pNZT-F | tacctatcacctcaaatggttcgc |
| pNZT-R | ctctagaggatcccaccgcg |
| sgRNA-F | cgcggtgggatcctctagagttttgagtgatcttctcaaaaaatactacc |
| sgRNA-R | accatttgaggtgataggtaaaaaaaagcaccgactcggtg |
| xylA-F | tcctctagagatatcgtcgaccgttttgggattttatcaacaatc |
| xylA-R | cttatccattccgatctcccccttcactt |
| Cas9-F | gggagatcggaatggataagaaatactcaataggcttaga |
| Cas9-R | cttgcatgcctgcaggtcgactcagtcacctcctagctgactcaa |
| HA1-F | attctgcagcagcggcggca |
| HA1-R | gcttaaaccatgtttggacagaattgatgacaaccggctc |
| HA2-F | gagccggttgtcatcaattctgtccaaacatggtttaagc |
| HA2-R | gaattgatgacaaccggctcc |
| HASOE-F | atactcgagattctgcagcagcggcggca |
| HASOE-R | atcctcgaggaattgatgacaaccggctc |
| diag-F | atgaaacaacaaaaacggct |
| diag-R | ctatctttgaacataaatt |
| Strains | Maximum Specific Growth Rates (h−1) 1 |
|---|---|
| BLG110 | 0.54 ± 0.02 |
| BLG107-induced | 0.49 ± 0.03 |
| BLG108-induced | 0.49 ± 0.01 |
| BLG109-induced | 0.60 ± 0.02 |
| BLG107-uninduced | 1.32 ± 0.05 |
| BLG108-uninduced | 1.40 ± 0.04 |
| BLG109-uninduced | 1.36 ± 0.06 |
| BLG100 | 1.21 ± 0.03 |
| Cultivations | T (°C) | Relative Expression of the cas9 Gene | Colonies on TB Agar | Colonies on Selective TB Agar | Target Gene Edited |
|---|---|---|---|---|---|
| ROUND 1 0.5% xylose for 24 h | 20 | 5.12 ± 0.11 | 30 ± 1 | 30 ± 1 | 2 ± 1 |
| 25 | 8.63 ± 0.35 | 31 ± 2 | 32 ± 1 | 1 ± 1 | |
| 30 | 15.92 ± 0.42 | 33 ± 2 | 33 ± 2 | 3 ± 1 | |
| 33 | 16.26 ± 0.55 | 34 ± 1 | 27 ± 1 | 1 ± 1 | |
| 37 | 7.9 1± 0.28 | 33 ± 1 | 4 ± 1 | 0 ± 1 | |
| 42 | 3.37 ± 0.12 | 32 ± 1 | 0 | 0 | |
| 50 | 0 | 30 ± 1 | 0 | 0 | |
| ROUND 2 0.5% xylose for 24 h | 20 | 3.32 ± 0.09 | 34 ± 1 | 34 ± 1 | 33 ± 1 |
| 25 | 5.57 ± 0.22 | 37 ± 1 | 37 ± 2 | 30 ± 1 | |
| 30 | 10.13 ± 0.52 | 39 ± 2 | 39 ± 2 | 26 ± 1 | |
| 33 | 10.91 ± 0.37 | 44 ± 2 | 44 ± 2 | 27 ± 1 | |
| 37 | 5.32 ± 0.20 | 41 ± 1 | 2 ± 1 | 12 ± 1 | |
| 42 | 0 | 38 ± 1 | 0 | 1 | |
| 50 | 0 | 38 ± 1 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, H.; Zhang, L.; Ding, Z.; Xu, S.; Gu, Z.; Shi, G. Efficient Genome Editing in Bacillus licheniformis Mediated by a Conditional CRISPR/Cas9 System. Microorganisms 2020, 8, 754. https://doi.org/10.3390/microorganisms8050754
Li Y, Wang H, Zhang L, Ding Z, Xu S, Gu Z, Shi G. Efficient Genome Editing in Bacillus licheniformis Mediated by a Conditional CRISPR/Cas9 System. Microorganisms. 2020; 8(5):754. https://doi.org/10.3390/microorganisms8050754
Chicago/Turabian StyleLi, Youran, Hanrong Wang, Liang Zhang, Zhongyang Ding, Sha Xu, Zhenghua Gu, and Guiyang Shi. 2020. "Efficient Genome Editing in Bacillus licheniformis Mediated by a Conditional CRISPR/Cas9 System" Microorganisms 8, no. 5: 754. https://doi.org/10.3390/microorganisms8050754