Environmental Impact on Differential Composition of Gut Microbiota in Indoor Chickens in Commercial Production and Outdoor, Backyard Chickens

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Sequencing of V3/V4 Region of 16S rRNA Genes
2.3. Real-Time PCR Quantification of Antibiotic Resistance Genes and Genes Specific for Representatives of Selected Genera
2.4. Statistics
3. Results
3.1. Microbiota Composition of Hens from Indoor/Commercial and Outdoor/Backyard Poultry Flocks
3.2. Real-Time PCR Quantification of Selected Genera
3.3. Real-Time PCR Quantification of Antibiotic Resistance Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kubasova, T.; Kollarcikova, M.; Crhanova, M.; Karasova, D.; Cejkova, D.; Sebkova, A.; Matiasovicova, J.; Faldynova, M.; Pokorna, A.; Cizek, A.; et al. Contact with adult hen affects development of caecal microbiota in newly hatched chicks. PLoS ONE 2019, 14, e0212446. [Google Scholar] [CrossRef] [PubMed]
- Rantala, M.; Nurmi, E. Prevention of the growth of Salmonella infantis in chicks by the flora of the alimentary tract of chickens. Br. Poult. Sci. 1973, 14, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Hofacre, C.L.; Froyman, R.; Gautrias, B.; George, B.; Goodwin, M.A.; Brown, J. Use of Aviguard and other intestinal bioproducts in experimental Clostridium perfringens-associated necrotizing enteritis in broiler chickens. Avian Dis. 1998, 42, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Craven, S.E.; Stern, N.J.; Cox, N.A.; Bailey, J.S.; Berrang, M. Cecal carriage of Clostridium perfringens in broiler chickens given Mucosal Starter Culture. Avian Dis. 1999, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Varmuzova, K.; Kubasova, T.; Davidova-Gerzova, L.; Sisak, F.; Havlickova, H.; Sebkova, A.; Faldynova, M.; Rychlik, I. Composition of gut microbiota influences resistance of newly hatched chickens to Salmonella Enteritidis Infection. Front. Microbiol. 2016, 7, 957. [Google Scholar] [CrossRef] [Green Version]
- Caly, D.L.; D′Inca, R.; Auclair, E.; Drider, D. Alternatives to antibiotics to prevent necrotic enteritis in broiler chickens: A microbiologist′s perspective. Front. Microbiol. 2015, 6, 1336. [Google Scholar] [CrossRef] [Green Version]
- Rychlik, I. Composition and function of chicken gut microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Videnska, P.; Sedlar, K.; Lukac, M.; Faldynova, M.; Gerzova, L.; Cejkova, D.; Sisak, F.; Rychlik, I. Succession and replacement of bacterial populations in the caecum of egg laying hens over their whole life. PLoS ONE 2014, 9, e115142. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Shi, S.; Tu, J.; Li, S. Comparative metagenomic sequencing analysis of cecum microbiotal diversity and function in broilers and layers. 3 Biotech 2019, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Ocejo, M.; Oporto, B.; Hurtado, A. 16S rRNA amplicon sequencing characterization of caecal microbiome composition of broilers and free-range slow-growing chickens throughout their productive lifespan. Sci. Rep. 2019, 9, 2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeant, M.J.; Constantinidou, C.; Cogan, T.A.; Bedford, M.R.; Penn, C.W.; Pallen, M.J. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS ONE 2014, 9, e91941. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, H.; Zhang, L.; Su, Y.; Shi, D.; Xiao, H.; Tian, Y. High-throughput sequencing technology to reveal the composition and function of cecal microbiota in Dagu chicken. BMC Microbiol. 2016, 16, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothrock, M.J., Jr.; Locatelli, A.; Feye, K.M.; Caudill, A.J.; Guard, J.; Hiett, K.; Ricke, S.C. A Microbiomic Analysis of a pasture-raised broiler flock elucidates foodborne pathogen ecology along the farm-to-fork continuum. Front. Vet. Sci. 2019, 6, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.; Alessandri, G.; Mancabelli, L.; Gering, E.; Mangifesta, M.; Milani, C.; Lugli, G.A.; Viappiani, A.; Duranti, S.; Turroni, F.; et al. Untangling the cecal microbiota of feral chickens by culturomic and metagenomic analyses. Environ. Microbiol. 2017, 19, 4771–4783. [Google Scholar] [CrossRef]
- Stanley, D.; Hughes, R.J.; Geier, M.S.; Moore, R.J. Bacteria within the Gastrointestinal Tract Microbiota correlated with improved growth and feed conversion: Challenges presented for the identification of performance enhancing probiotic bacteria. Front. Microbiol. 2016, 7, 187. [Google Scholar] [CrossRef] [Green Version]
- Stanley, D.; Geier, M.S.; Chen, H.; Hughes, R.J.; Moore, R.J. Comparison of fecal and cecal microbiotas reveals qualitative similarities but quantitative differences. BMC Microbiol 2015, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Willson, N.L.; Nattrass, G.S.; Hughes, R.J.; Moore, R.J.; Stanley, D.; Hynd, P.I.; Forder, R.E.A. Correlations between intestinal innate immune genes and cecal microbiota highlight potential for probiotic development for immune modulation in poultry. Appl. Microbiol. Biotechnol. 2018, 102, 9317–9329. [Google Scholar] [CrossRef]
- Medvecky, M.; Cejkova, D.; Polansky, O.; Karasova, D.; Kubasova, T.; Cizek, A.; Rychlik, I. Whole genome sequencing and function prediction of 133 gut anaerobes isolated from chicken caecum in pure cultures. BMC Genomics 2018, 19, 561. [Google Scholar] [CrossRef] [Green Version]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef] [PubMed]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, G.; Shoemaker, N.B.; Salyers, A.A. The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell Mol. Life Sci. 2002, 59, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczak, K.A.; Flint, H.J.; Scott, K.P. Comparative analysis of sequences flanking tet(W) resistance genes in multiple species of gut bacteria. Antimicrob. Agents Chemother. 2006, 50, 2632–2639. [Google Scholar] [CrossRef] [Green Version]
- Ammor, M.S.; Florez, A.B.; van Hoek, A.H.; de Los Reyes-Gavilan, C.G.; Aarts, H.J.; Margolles, A.; Mayo, B. Molecular characterization of intrinsic and acquired antibiotic resistance in lactic acid bacteria and bifidobacteria. J. Mol. Microbiol. Biotechnol. 2008, 14, 6–15. [Google Scholar] [CrossRef]
- Melville, C.M.; Scott, K.P.; Mercer, D.K.; Flint, H.J. Novel tetracycline resistance gene, tet(32), in the Clostridium-related human colonic anaerobe K10 and its transmission in vitro to the rumen anaerobe Butyrivibrio fibrisolvens. Antimicrob. Agents Chemother. 2001, 45, 3246–3249. [Google Scholar] [CrossRef] [Green Version]
- Naito, M.; Sato, K.; Shoji, M.; Yukitake, H.; Ogura, Y.; Hayashi, T.; Nakayama, K. Characterization of the Porphyromonas gingivalis conjugative transposon CTnPg1: Determination of the integration site and the genes essential for conjugal transfer. Microbiology 2011, 157, 2022–2032. [Google Scholar] [CrossRef] [Green Version]
- Buckwold, S.L.; Shoemaker, N.B.; Sears, C.L.; Franco, A.A. Identification and characterization of conjugative transposons CTn86 and CTn9343 in Bacteroides fragilis strains. Appl. Environ. Microbiol. 2007, 73, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Schmid, S.; Bevilacqua, C.; Crutz-Le Coq, A.M. Alternative sigma factor sigmaH activates competence gene expression in Lactobacillus sakei. BMC Microbiol. 2012, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Inniss, N.L.; Prehna, G.; Morrison, D.A. The pneumococcal sigma(X) activator, ComW, is a DNA-binding protein critical for natural transformation. J. Biol. Chem. 2019, 294, 11101–11118. [Google Scholar] [CrossRef]
- Kubasova, T.; Kollarcikova, M.; Crhanova, M.; Karasova, D.; Cejkova, D.; Sebkova, A.; Matiasovicova, J.; Faldynova, M.; Sisak, F.; Babak, V.; et al. Gut anaerobes capable of chicken caecum colonisation. Microorganisms 2019, 7, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]


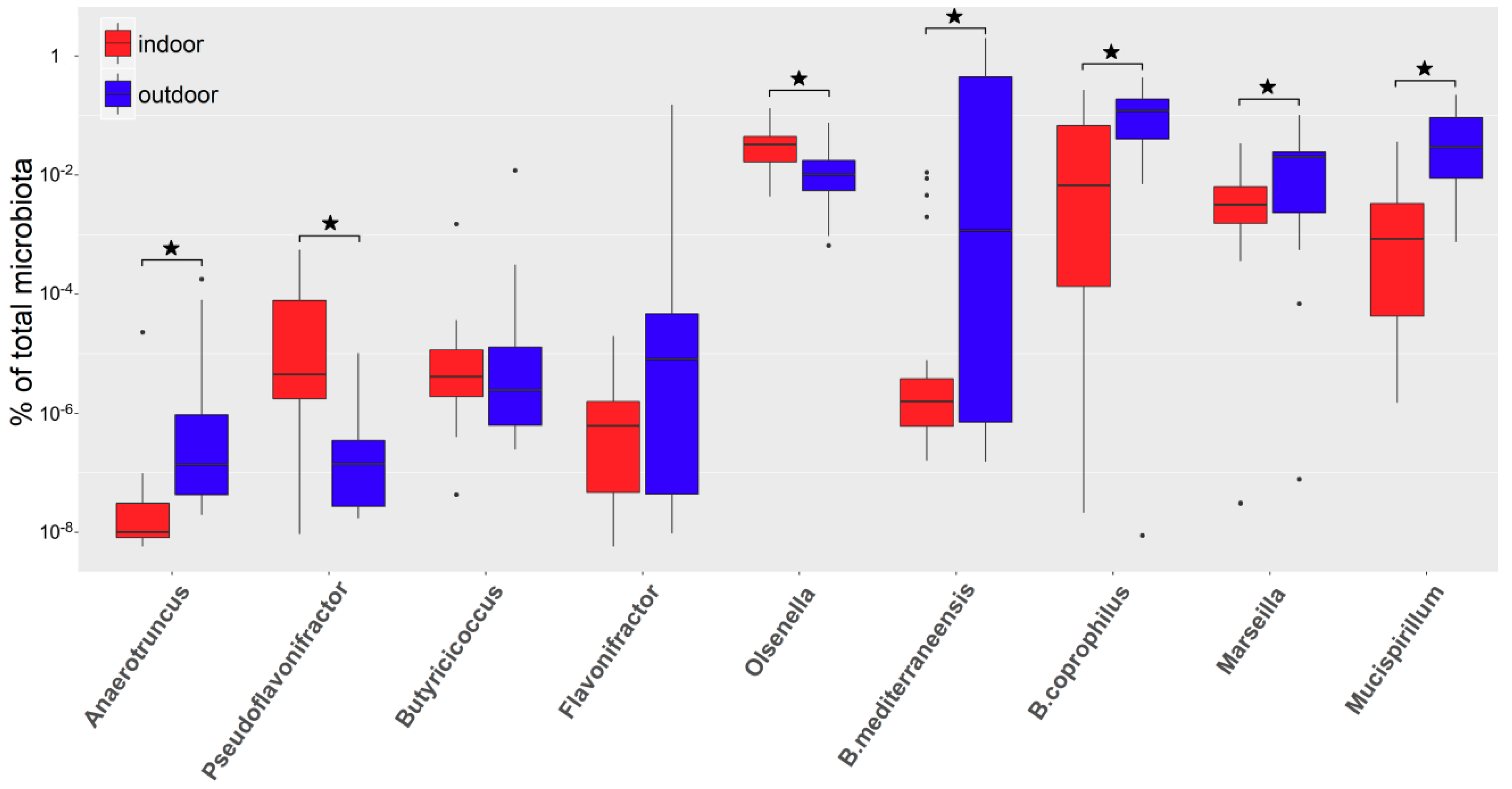
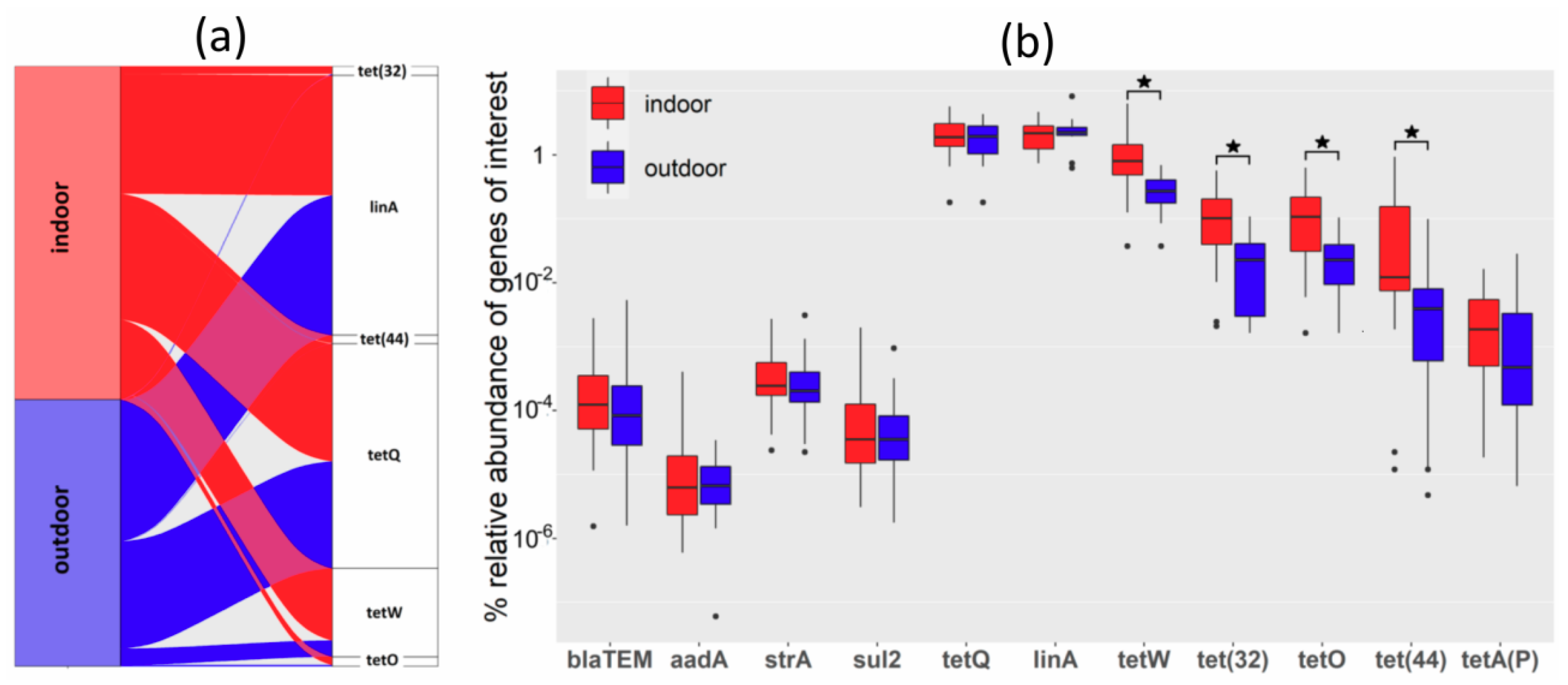

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Genus | In/Out # | Indoor * | Outdoor * | p Value |
|---|---|---|---|---|---|
| Actinobacteria | Olsenella$ | 2.59 | 0.71 | 0.27 | p < 0.01 |
| Bacteroidetes | Tannerella | 2.94 | 0.40 | 0.14 | p < 0.01 |
| Bacteroidetes | Flavobacteriaceae;Other | 2.55 | 0.45 | 0.18 | p < 0.01 |
| Bacteroidetes | Phocaeicola | 2.49 | 0.17 | 0.07 | p < 0.01 |
| Bacteroidetes | Odoribacter | 1.85 | 0.49 | 0.27 | p < 0.01 |
| Bacteroidetes | Bacteroidetes;Other | 1.66 | 8.53 | 5.13 | p < 0.01 |
| Bacteroidetes | Porphyromonadaceae;Other | 1.50 | 3.63 | 2.42 | p < 0.01 |
| Bacteroidetes | Bacteroidales;Other | 1.46 | 7.35 | 05.3 | p < 0.05 |
| Candidatus Saccharibacteria | Saccharibacteria_genera_incertae_sedis | 5.15 | 0.51 | 0.10 | p < 0.01 |
| Euryarchaeota | Methanobrevibacter | 4.92 | 0.19 | 0.04 | p < 0.01 |
| Firmicutes | Acetanaerobacterium | 6.29 | 0.19 | 0.03 | p < 0.01 |
| Firmicutes | Ethanoligenens | 5.64 | 0.10 | 0.02 | p < 0.01 |
| Firmicutes | Anaerotruncus | 04.12 | 0.25 | 0.06 | p < 0.01 |
| Firmicutes | Peptococcus | 3.60 | 0.13 | 0.04 | p < 0.01 |
| Firmicutes | Firmicutes;Other | 3.35 | 0.37 | 0.11 | p < 0.01 |
| Firmicutes | Acidaminococcaceae;Other | 3.28 | 0.12 | 0.04 | p < 0.01 |
| Firmicutes | Coprococcus | 03.7 | 0.14 | 0.04 | p < 0.01 |
| Firmicutes | Eubacterium | 3.00 | 0.12 | 0.04 | p < 0.01 |
| Firmicutes | Flavonifractor | 2.94 | 1.17 | 0.40 | p < 0.01 |
| Firmicutes | Ruminococcus2 | 2.81 | 2.67 | 0.95 | p < 0.01 |
| Firmicutes | Pseudoflavonifractor | 2.77 | 0.95 | 0.34 | p < 0.01 |
| Firmicutes | Clostridium XI | 2.63 | 0.32 | 0.12 | p < 0.01 |
| Firmicutes | Clostridium IV | 2.56 | 0.72 | 0.28 | p < 0.01 |
| Firmicutes | Ruminococcus | 2.54 | 0.15 | 0.06 | p < 0.01 |
| Firmicutes | Lachnospiraceae;Other | 2.47 | 2.98 | 1.20 | p < 0.01 |
| Firmicutes | Ruminococcaceae;Other | 2.41 | 4.49 | 1.86 | p < 0.01 |
| Firmicutes | Butyricicoccus | 2.38 | 0.41 | 0.17 | p < 0.01 |
| Firmicutes | Roseburia | 2.27 | 0.16 | 0.07 | p < 0.01 |
| Firmicutes | Erysipelotrichaceae_incertae_sedis | 02.5 | 0.14 | 0.07 | p < 0.05 |
| Firmicutes | Subdoligranulum | 1.90 | 0.46 | 0.24 | p < 0.01 |
| Firmicutes | Oscillibacter | 1.89 | 1.25 | 0.66 | p < 0.01 |
| Firmicutes | Clostridiales;Other | 1.78 | 3.39 | 1.90 | p < 0.05 |
| Firmicutes | Blautia | 1.77 | 0.27 | 0.15 | p < 0.05 |
| Firmicutes | Lachnospiracea_incertae_sedis | 1.54 | 0.13 | 0.08 | p < 0.05 |
| Firmicutes | Clostridium XlVa | 1.43 | 0.92 | 0.64 | p < 0.05 |
| Firmicutes | Clostridium XlVb | 1.38 | 0.36 | 0.26 | p < 0.05 |
| Firmicutes | Clostridia;Other | 1.26 | 0.12 | 0.09 | p < 0.05 |
| Proteobacteria | Vampirovibrio | 3.99 | 0.49 | 0.12 | p < 0.05 |
| Proteobacteria | Parasutterella | 1.46 | 0.43 | 0.29 | p < 0.05 |
| Spirochaetes | Spirochaetaceae;Other | 1.78 | 0.27 | 0.15 | p < 0.05 |
| Verrucomicrobia | Opitutae;Other | 199.31 | 0.38 | 0.00 | p < 0.01 |
| Verrucomicrobia | Subdivision5_genera_incertae_sedis | 9.65 | 01.1 | 0.10 | p < 0.01 |
| Verrucomicrobia | Akkermansia | 9.50 | 0.11 | 0.01 | p < 0.01 |
| Bacteroidetes | Bacteroides | 0.59 | 15.57 | 26.51 | p < 0.01 |
| Bacteroidetes | Prevotella | 0.25 | 02.4 | 08.9 | p < 0.05 |
| Deferribacteres | Mucispirillum | 0.29 | 0.20 | 0.69 | p < 0.01 |
| Firmicutes | Dialister | 0.04 | 0.04 | 01.5 | p < 0.01 |
| Lentisphaerae | Victivallis | 0.30 | 0.16 | 0.54 | p < 0.05 |
| Proteobacteria | Succinatimonas | 0.44 | 0.12 | 0.27 | p < 0.01 |
| Proteobacteria | Sutterellaceae;Other | 0.38 | 0.08 | 0.22 | p < 0.01 |
| Proteobacteria | Sutterella | 0.21 | 0.26 | 1.21 | p < 0.01 |
| Proteobacteria | Anaerobiospirillum | 0.13 | 0.29 | 2.32 | p < 0.01 |
| Tenericutes | Asteroleplasma | 0.26 | 0.15 | 0.59 | p < 0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidlerova, Z.; Kubasova, T.; Faldynova, M.; Crhanova, M.; Karasova, D.; Babak, V.; Rychlik, I. Environmental Impact on Differential Composition of Gut Microbiota in Indoor Chickens in Commercial Production and Outdoor, Backyard Chickens. Microorganisms 2020, 8, 767. https://doi.org/10.3390/microorganisms8050767
Seidlerova Z, Kubasova T, Faldynova M, Crhanova M, Karasova D, Babak V, Rychlik I. Environmental Impact on Differential Composition of Gut Microbiota in Indoor Chickens in Commercial Production and Outdoor, Backyard Chickens. Microorganisms. 2020; 8(5):767. https://doi.org/10.3390/microorganisms8050767
Chicago/Turabian StyleSeidlerova, Zuzana, Tereza Kubasova, Marcela Faldynova, Magdalena Crhanova, Daniela Karasova, Vladimir Babak, and Ivan Rychlik. 2020. "Environmental Impact on Differential Composition of Gut Microbiota in Indoor Chickens in Commercial Production and Outdoor, Backyard Chickens" Microorganisms 8, no. 5: 767. https://doi.org/10.3390/microorganisms8050767