C3 Opsonization of Anthrax Bacterium and Peptidoglycan Supports Recognition and Activation of Neutrophils

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analysis of Primary Human Neutrophils
2.3. Bacteria Strain and Peptidoglycan Purification
2.4. Neutrophil Recognition of hkBa and PGN
2.5. Neutrophil Phagocytosis of hkBa and PGN
2.6. Neutrophil Degranulation
2.7. MPO Immunoreactivity in Neutrophils
2.8. Data Analysis and Representation
3. Results
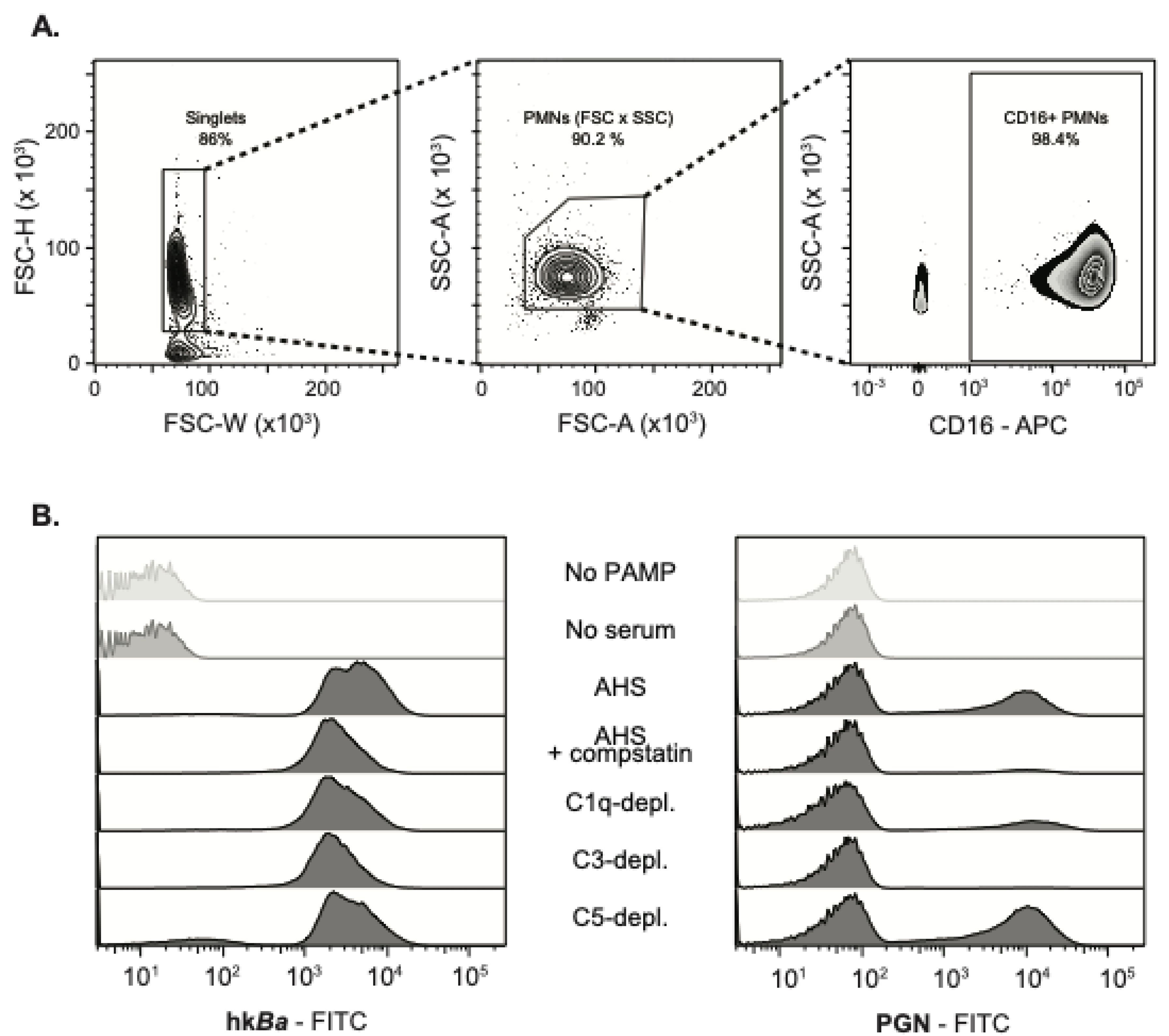
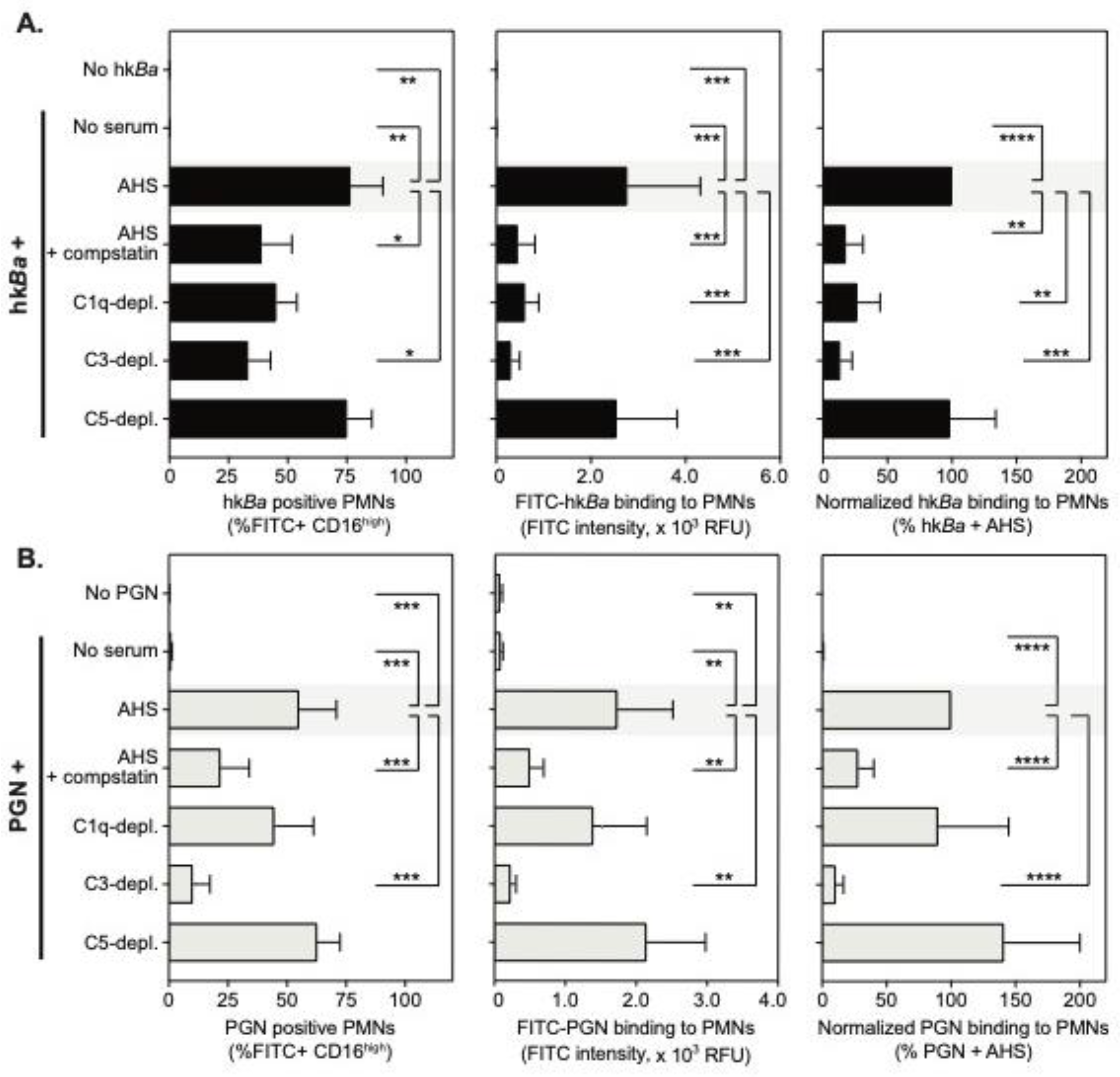
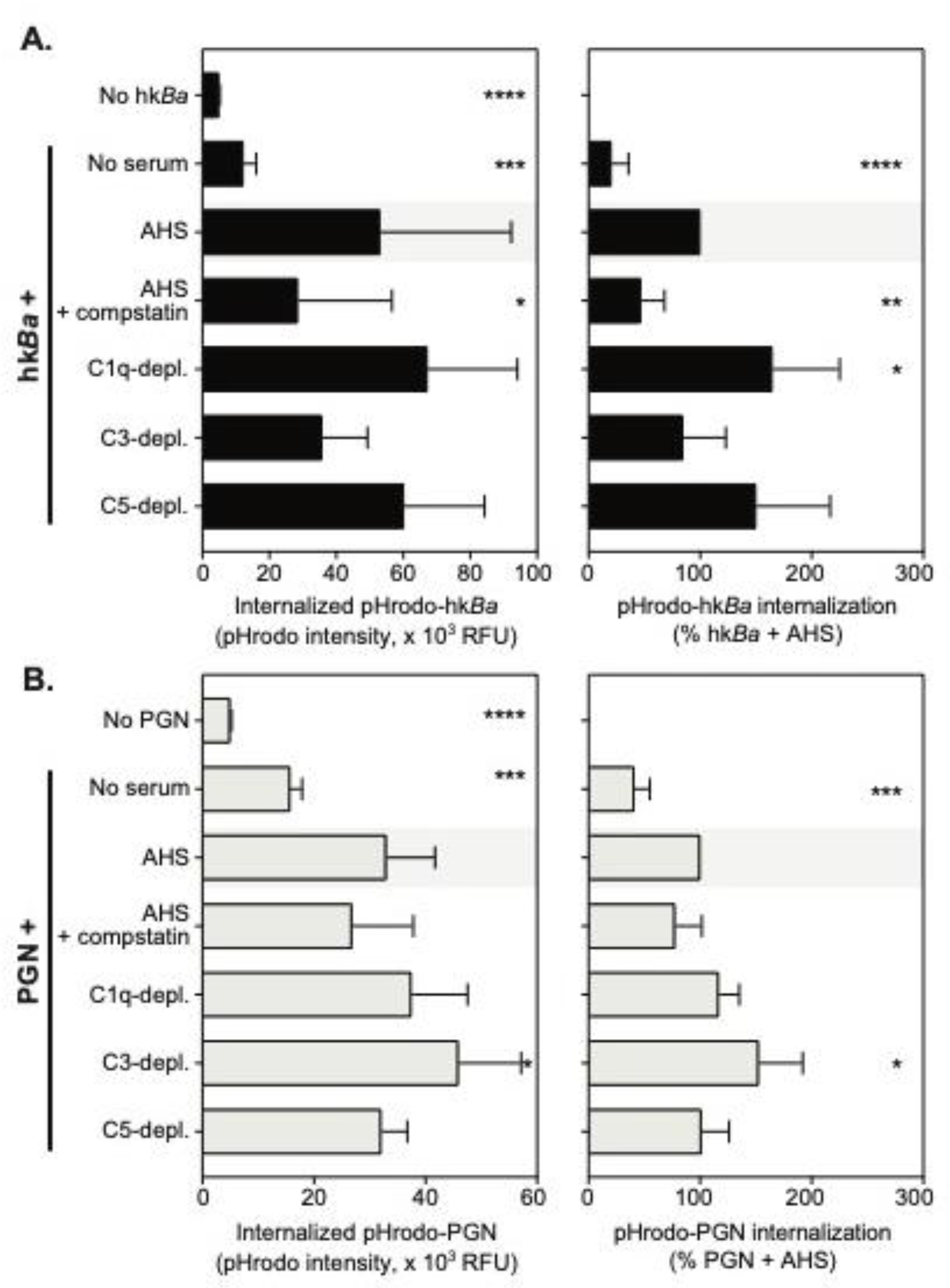
3.1. Complement-Dependent Recognition of Anthrax Bacterium and Peptidoglycan
3.2. C3b Opsonization Requirements for the Internalization of Anthrax Bacterium and Peptidoglycan
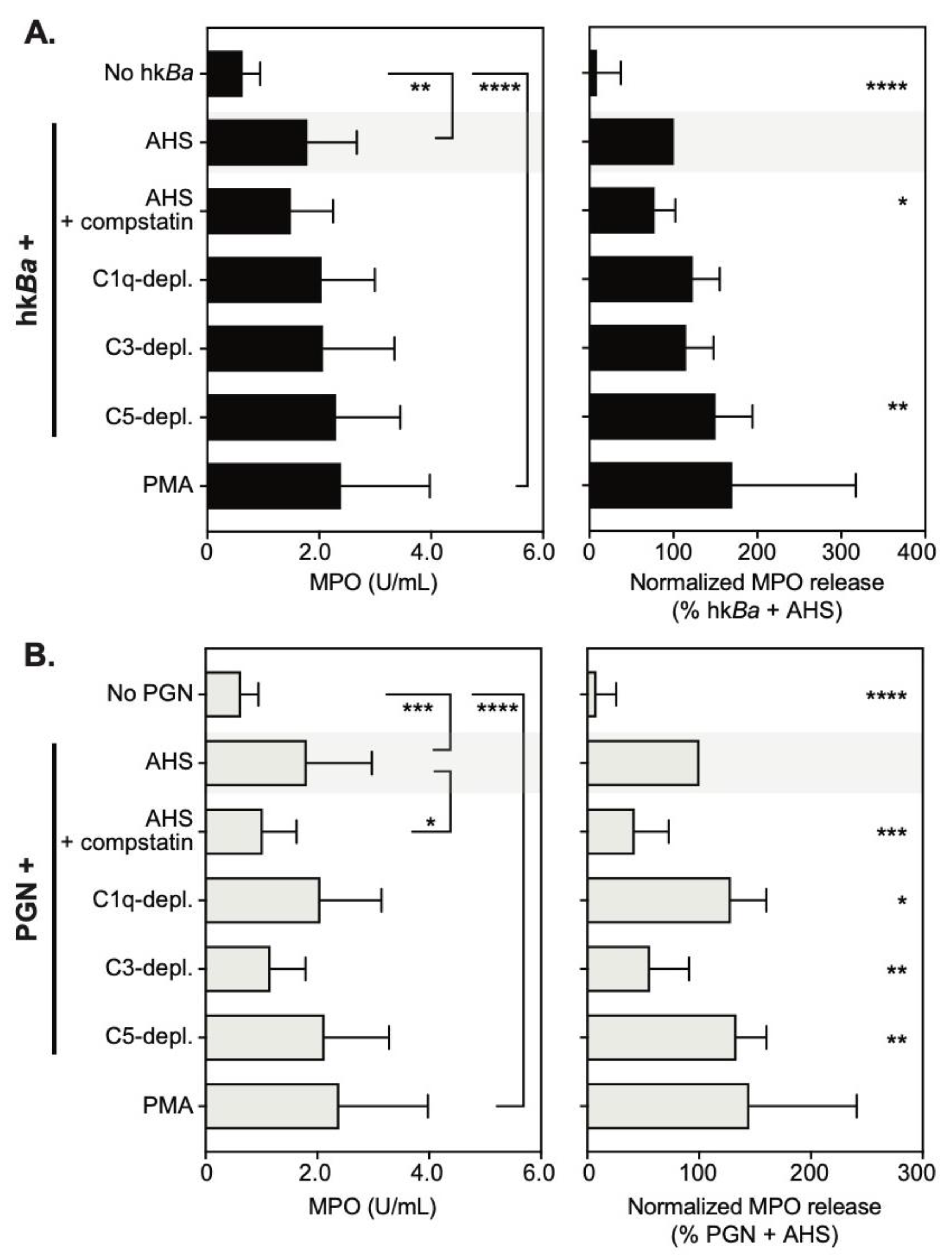
3.3. C3b Opsonization Requirements for Neutrophil Degranulation
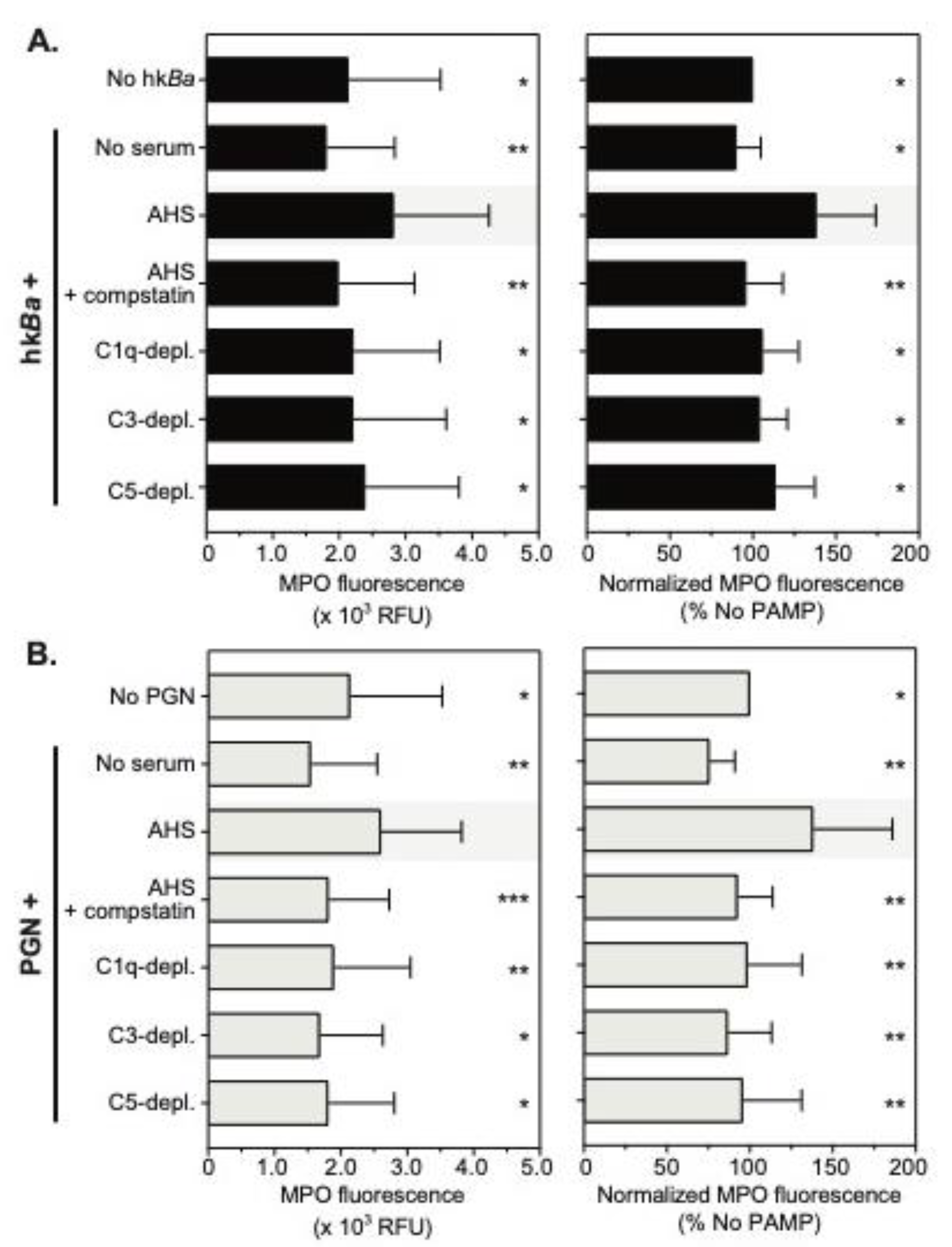
3.4. Complement Enhances Neutrophil MPO Immunoreactivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sweeney, D.A.; Hicks, C.W.; Cui, X.; Li, Y.; Eichacker, P.Q. Anthrax infection. Am. J. Respir. Crit. Care Med. 2011, 184, 1333–1341. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.J.; Kracalik, I.T.; Ross, N.; Alexander, K.A.; Hugh-Jones, M.E.; Fegan, M.; Elkin, B.T.; Epp, T.; Shury, T.K.; Zhang, W.; et al. The global distribution of Bacillus anthracis and associated anthrax risk to humans, livestock and wildlife. Nat. Microbiol. 2019, 4, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.G.; de Smalen, A.W.; Mor, S.M. Climatic influence on anthrax suitability in warming northern latitudes. Sci. Rep. 2018, 8, 9269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglesby, T.V.; O’Toole, T.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Friedlander, A.M.; Gerberding, J.; Hauer, J.; Hughes, J.; et al. Anthrax as a biological weapon, 2002: updated recommendations for management. JAMA 2002, 287, 2236–2252. [Google Scholar] [CrossRef]
- Meselson, M.; Guillemin, J.; Hugh-Jones, M.; Langmuir, A.; Popova, I.; Shelokov, A.; Yampolskaya, O. The Sverdlovsk anthrax outbreak of 1979. Science 1994, 266, 1202–1208. [Google Scholar] [CrossRef]
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar] [CrossRef]
- Coggeshall, K.M.; Lupu, F.; Ballard, J.; Metcalf, J.P.; James, J.A.; Farris, D.; Kurosawa, S. The sepsis model: an emerging hypothesis for the lethality of inhalation anthrax. J. Cell Mol. Med. 2013, 17, 914–920. [Google Scholar] [CrossRef]
- Goossens, P.L.; Tournier, J.N. Crossing of the epithelial barriers by Bacillus anthracis: the Known and the Unknown. Front. Microbiol. 2015, 6, 1122. [Google Scholar] [CrossRef] [Green Version]
- Premanandan, C.; Storozuk, C.A.; Clay, C.D.; Lairmore, M.D.; Schlesinger, L.S.; Phipps, A.J. Complement protein C3 binding to Bacillus anthracis spores enhances phagocytosis by human macrophages. Microb. Pathog. 2009, 46, 306–314. [Google Scholar] [CrossRef]
- Gu, C.; Jenkins, S.A.; Xue, Q.; Xu, Y. Activation of the classical complement pathway by Bacillus anthracis is the primary mechanism for spore phagocytosis and involves the spore surface protein BclA. J. Immunol. 2012, 188, 4421–4431. [Google Scholar] [CrossRef] [Green Version]
- Oliva, C.R.; Swiecki, M.K.; Griguer, C.E.; Lisanby, M.W.; Bullard, D.C.; Turnbough, C.L., Jr.; Kearney, J.F. The integrin Mac-1 (CR3) mediates internalization and directs Bacillus anthracis spores into professional phagocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 1261–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, T.C.; Fadl, A.A.; Koehler, T.M.; Swanson, J.A.; Hanna, P.C. Early Bacillus anthracis-macrophage interactions: intracellular survival survival and escape. Cell Microbiol. 2000, 2, 453–463. [Google Scholar] [CrossRef]
- Shetron-Rama, L.M.; Herring-Palmer, A.C.; Huffnagle, G.B.; Hanna, P. Transport of Bacillus anthracis from the lungs to the draining lymph nodes is a rapid process facilitated by CD11c+ cells. Microb. Pathog. 2010, 49, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009, 30, 439–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candela, T.; Fouet, A. Poly-gamma-glutamate in bacteria. Mol. Microbiol. 2006, 60, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Demirdag, K.; Ozden, M.; Saral, Y.; Kalkan, A.; Kilic, S.S.; Ozdarendeli, A. Cutaneous anthrax in adults: a review of 25 cases in the eastern Anatolian region of Turkey. Infection 2003, 31, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.L.; Bischof, T.S.; Sohnle, P.G. Superficial exudates of neutrophils prevent invasion of Bacillus anthracis bacilli into abraded skin of resistant mice. Int, J. Exp. Pathol. 2008, 89, 180–187. [Google Scholar] [CrossRef]
- Cote, C.K.; Van Rooijen, N.; Welkos, S.L. Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect. Immun. 2006, 74, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; Ali, S.R.; Bier, E.; Nizet, V. Innate Immune Interactions between Bacillus anthracis and Host Neutrophils. Front. Cell Infect. Microbiol. 2018, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Moayeri, M.; Crown, D.; Newman, Z.L.; Okugawa, S.; Eckhaus, M.; Cataisson, C.; Liu, S.; Sastalla, I.; Leppla, S.H. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 2010, 6, e1001222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns-Kurosawa, D.J.; Lupu, F.; Taylor, F.B., Jr.; Kinasewitz, G.; Kurosawa, S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am. J. Pathol. 2006, 169, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Scholl, A.; Hurwitz, R.; Brinkmann, V.; Schmid, M.; Jungblut, P.; Weinrauch, Y.; Zychlinsky, A. Human neutrophils kill Bacillus anthracis. PLoS Pathog. 2005, 1, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg, L.M.; Abramova, F.A.; Yampolskaya, O.V.; Walker, D.H.; Smith, J.H. Quantitative pathology of inhalational anthrax I: quantitative microscopic findings. Mod. Pathol. 2001, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Sanz, P.; Teel, L.D.; Alem, F.; Carvalho, H.M.; Darnell, S.C.; O’Brien, A.D. Detection of Bacillus anthracis spore germination in vivo by bioluminescence imaging. Infect. Immun. 2008, 76, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Crawford, M.A.; Aylott, C.V.; Bourdeau, R.W.; Bokoch, G.M. Bacillus anthracis toxins inhibit human neutrophil NADPH oxidase activity. J. Immunol. 2006, 176, 7557–7565. [Google Scholar] [CrossRef] [Green Version]
- Weiner, Z.P.; Ernst, S.M.; Boyer, A.E.; Gallegos-Candela, M.; Barr, J.R.; Glomski, I.J. Circulating lethal toxin decreases the ability of neutrophils to respond to Bacillus anthracis. Cell Microbiol. 2014, 16, 504–518. [Google Scholar] [CrossRef]
- Wang, Y.; Jonsson, F. Expression, Role, and Regulation of Neutrophil Fcgamma Receptors. Front. Immunol. 2019, 10, 1958. [Google Scholar] [CrossRef]
- Gasque, P. Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 2004, 41, 1089–1098. [Google Scholar] [CrossRef]
- Erdei, A.; Lukacsi, S.; Macsik-Valent, B.; Nagy-Balo, Z.; Kurucz, I.; Bajtay, Z. Non-identical twins: Different faces of CR3 and CR4 in myeloid and lymphoid cells of mice and men. Semin. Cell Dev. Biol. 2019, 85, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, F.; Means, T.K.; Luster, A.D. Toll-like receptors stimulate human neutrophil function. Blood 2003, 102, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Popescu, N.I.; Silasi, R.; Keshari, R.S.; Girton, A.; Burgett, T.; Zeerleder, S.S.; Gailani, D.; Gruber, A.; Lupu, F.; Coggeshall, K.M. Peptidoglycan induces disseminated intravascular coagulation in baboons through activation of both coagulation pathways. Blood 2018, 132, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, P.; Li, Y.; Shiloach, J.; Cui, X.; Sun, J.; Trinh, L.; Kubler-Kielb, J.; Vinogradov, E.; Mani, H.; Al-Hamad, M.; et al. Bacillus anthracis cell wall peptidoglycan but not lethal or edema toxins produces changes consistent with disseminated intravascular coagulation in a rat model. J. Infect. Dis. 2013, 208, 978–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popescu, N.I.; Girton, A.; Burgett, T.; Lovelady, K.; Coggeshall, K.M. Monocyte procoagulant responses to anthrax peptidoglycan are reinforced by proinflammatory cytokine signaling. Blood Adv. 2019, 3, 2436–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, J.K.; Khurana, T.; Langer, M.; West, C.M.; Ballard, J.D.; Metcalf, J.P.; Merkel, T.J.; Coggeshall, K.M. Inflammatory cytokine response to Bacillus anthracis peptidoglycan requires phagocytosis and lysosomal trafficking. Infect. Immun. 2010, 78, 2418–2428. [Google Scholar] [CrossRef] [Green Version]
- Langer, M.; Girton, A.W.; Popescu, N.I.; Burgett, T.; Metcalf, J.P.; Coggeshall, K.M. Neither Lys- and DAP-type peptidoglycans stimulate mouse or human innate immune cells via Toll-like receptor 2. PLoS ONE 2018, 13, e0193207. [Google Scholar] [CrossRef] [Green Version]
- Langer, M.; Malykhin, A.; Maeda, K.; Chakrabarty, K.; Williamson, K.S.; Feasley, C.L.; West, C.M.; Metcalf, J.P.; Coggeshall, K.M. Bacillus anthracis peptidoglycan stimulates an inflammatory response in monocytes through the p38 mitogen-activated protein kinase pathway. PLoS ONE 2008, 3, e3706. [Google Scholar] [CrossRef]
- Girton, A.W.; Popescu, N.I.; Keshari, R.S.; Burgett, T.; Lupu, F.; Coggeshall, K.M. Serum Amyloid P and IgG Exhibit Differential Capabilities in the Activation of the Innate Immune System in Response to Bacillus anthracis Peptidoglycan. Infect. Immun. 2018, 86, e00076-18. [Google Scholar] [CrossRef] [Green Version]
- Pulli, B.; Ali, M.; Forghani, R.; Schob, S.; Hsieh, K.L.; Wojtkiewicz, G.; Linnoila, J.J.; Chen, J.W. Measuring myeloperoxidase activity in biological samples. PLoS One 2013, 8, e67976. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.J.; Halff, E.F.; Lambris, J.D.; Gros, P. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J. Biol. Chem. 2007, 282, 29241–29247. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Popescu, N.I.; Raisley, B.; Keshari, R.S.; Dale, G.L.; Lupu, F.; Coggeshall, K.M. Bacillus anthracis peptidoglycan activates human platelets through FcgammaRII and complement. Blood 2013, 122, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Borregaard, N.; Cowland, J.B. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 1997, 89, 3503–3521. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, S.; Moretti, S.; Perruccio, K.; Fallarino, F.; Bozza, S.; Montagnoli, C.; Mosci, P.; Lipford, G.B.; Pitzurra, L.; Romani, L. TLRs govern neutrophil activity in aspergillosis. J. Immunol. 2004, 173, 7406–7415. [Google Scholar] [CrossRef]
- Cleret, A.; Quesnel-Hellmann, A.; Vallon-Eberhard, A.; Verrier, B.; Jung, S.; Vidal, D.; Mathieu, J.; Tournier, J.N. Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J. Immunol. 2007, 178, 7994–8001. [Google Scholar] [CrossRef] [Green Version]
- Bredius, R.G.; Fijen, C.A.; De Haas, M.; Kuijper, E.J.; Weening, R.S.; Van de Winkel, J.G.; Out, T.A. Role of neutrophil Fc gamma RIIa (CD32) and Fc gamma RIIIb (CD16) polymorphic forms in phagocytosis of human IgG1- and IgG3-opsonized bacteria and erythrocytes. Immunology 1994, 83, 624–630. [Google Scholar]
- Fallman, M.; Andersson, R.; Andersson, T. Signaling properties of CR3 (CD11b/CD18) and CR1 (CD35) in relation to phagocytosis of complement-opsonized particles. J. Immunol. 1993, 151, 330–338. [Google Scholar] [PubMed]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood 2012, 120, 4421–4431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, C.; Arve, S.; Ledbetter, J.; Elkon, K.B. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J. Exp. Med. 2017, 214, 2103–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Raisley, B.; Langer, M.; Iyer, J.K.; Vedham, V.; Ballard, J.L.; James, J.A.; Metcalf, J.; Coggeshall, K.M. Anti-peptidoglycan antibodies and Fcgamma receptors are the key mediators of inflammation in Gram-positive sepsis. J. Immunol. 2012, 189, 2423–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.O.; Mallin, R.L.; Krych-Goldberg, M.; Wang, X.; Hauhart, R.E.; Bromek, K.; Uhrin, D.; Atkinson, J.P.; Barlow, P.N. Structure of the C3b binding site of CR1 (CD35), the immune adherence receptor. Cell 2002, 108, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.K.; Bajic, G.; Sen, M.; Springer, T.A.; Vorup-Jensen, T.; Andersen, G.R. Complement receptor 3 forms a compact high affinity complex with iC3b. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Maenaka, K.; van der Merwe, P.A.; Stuart, D.I.; Jones, E.Y.; Sondermann, P. The human low affinity Fcgamma receptors IIa, IIb, and III bind IgG with fast kinetics and distinct thermodynamic properties. J. Biol. Chem. 2001, 276, 44898–44904. [Google Scholar] [CrossRef] [Green Version]
- Myones, B.L.; Dalzell, J.G.; Hogg, N.; Ross, G.D. Neutrophil and monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3. J. Clin. Invest. 1988, 82, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Klickstein, L.B.; Barbashov, S.F.; Liu, T.; Jack, R.M.; Nicholson-Weller, A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity 1997, 7, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Boesch, A.W.; Brown, E.P.; Cheng, H.D.; Ofori, M.O.; Normandin, E.; Nigrovic, P.A.; Alter, G.; Ackerman, M.E. Highly parallel characterization of IgG Fc binding interactions. MAbs 2014, 6, 915–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, J.K.; Coggeshall, K.M. Cutting edge: primary innate immune cells respond efficiently to polymeric peptidoglycan, but not to peptidoglycan monomers. J. Immunol. 2011, 186, 3841–3845. [Google Scholar] [CrossRef] [Green Version]
- Vetvicka, V.; Thornton, B.P.; Ross, G.D. Soluble beta-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J. Clin. Invest. 1996, 98, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Thornton, B.P.; Vetvicka, V.; Pitman, M.; Goldman, R.C.; Ross, G.D. Analysis of the sugar specificity and molecular location of the beta-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18). J. Immunol. 1996, 156, 1235–1246. [Google Scholar] [PubMed]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popescu, N.I.; Keshari, R.S.; Cochran, J.; Coggeshall, K.M.; Lupu, F. C3 Opsonization of Anthrax Bacterium and Peptidoglycan Supports Recognition and Activation of Neutrophils. Microorganisms 2020, 8, 1039. https://doi.org/10.3390/microorganisms8071039
Popescu NI, Keshari RS, Cochran J, Coggeshall KM, Lupu F. C3 Opsonization of Anthrax Bacterium and Peptidoglycan Supports Recognition and Activation of Neutrophils. Microorganisms. 2020; 8(7):1039. https://doi.org/10.3390/microorganisms8071039
Chicago/Turabian StylePopescu, Narcis I., Ravi S. Keshari, Jackie Cochran, K. Mark Coggeshall, and Florea Lupu. 2020. "C3 Opsonization of Anthrax Bacterium and Peptidoglycan Supports Recognition and Activation of Neutrophils" Microorganisms 8, no. 7: 1039. https://doi.org/10.3390/microorganisms8071039