Can We Harness Immune Responses to Improve Drug Treatment in Leishmaniasis?

 , , and
, , and
Abstract
:1. Introduction
2. Benchmark Drugs for Leishmaniasis
2.1. Pentavalent Antimonials
2.1.1. Dosage and Side-Effects
2.1.2. Mechanism of Action and Immune Response
2.1.3. Antimony Resistance
2.2. Amphotericin B
2.2.1. Dosage and Side-Effects
2.2.2. Mechanism of Action and Immune Response
2.2.3. AmB Resistance
2.3. Paromomycin
2.3.1. Dosage and Side-Effects
2.3.2. Mechanism of Action and Immune Response
2.3.3. PR Resistance
2.4. Miltefosine
2.4.1. Dosage and Side-Effects
2.4.2. Mechanism of Action and Immune Response
2.4.3. MIL Resistance
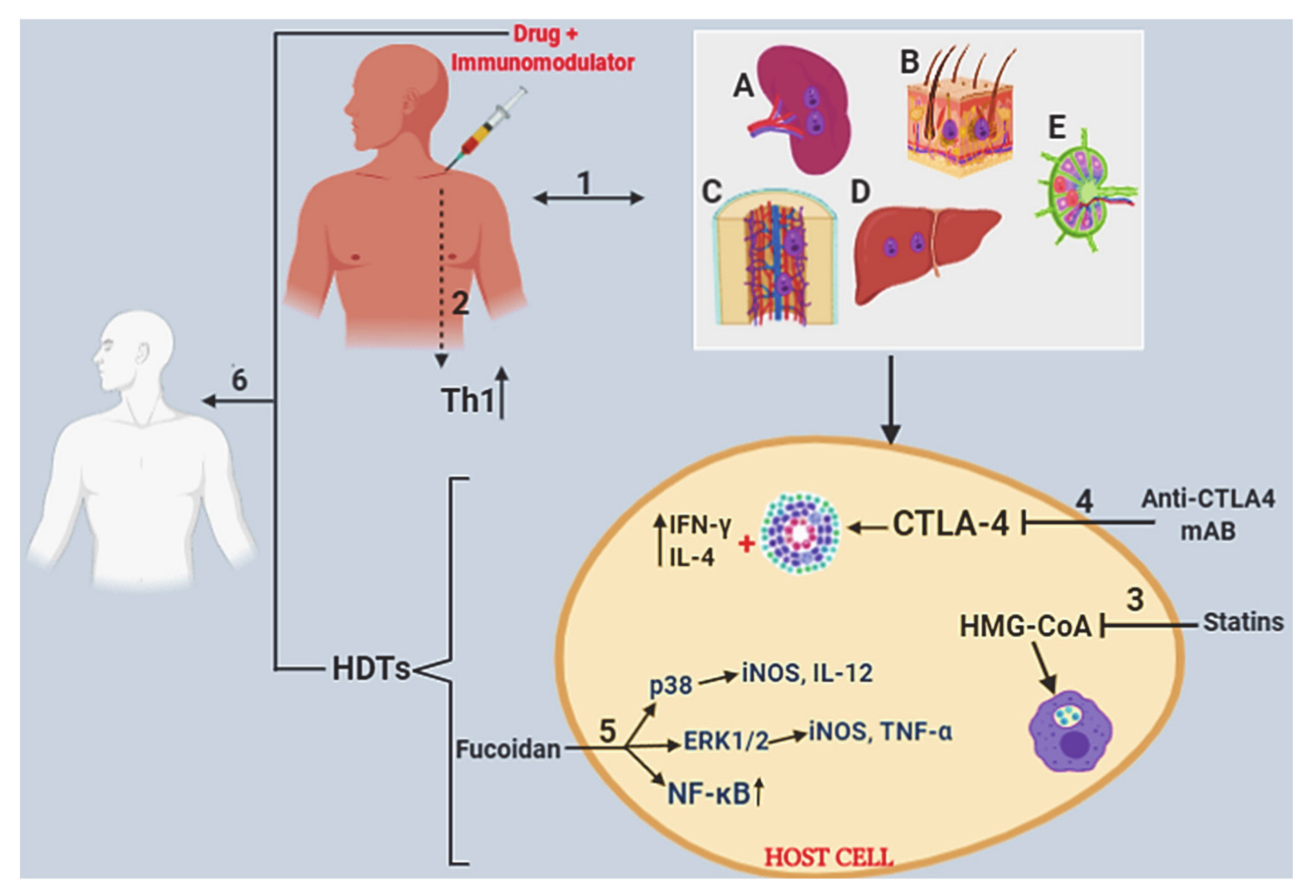
2.5. Host-Directed Therapeutics
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in Leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votypka, J.; Marty, P.; Delaunay, P.; Sereno, D. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef] [PubMed]
- Paranaiba, L.F.; Pinheiro, L.J.; Torrecilhas, A.C.; Macedo, D.H.; Menezes-Neto, A.; Tafuri, W.L.; Soares, R.P. Leishmania enriettii (Muniz & Medina, 1948): A highly diverse parasite is here to stay. PLoS Pathog. 2017, 13, e1006303. [Google Scholar]
- Steverding, D. The history of Leishmaniasis. Parasites Vectors 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittra, B.; Laranjeira-Silva, M.F.; Miguel, D.C.; Perrone Bezerra de Menezes, J.; Andrews, N.W. The iron-dependent mitochondrial superoxide dismutase SODA promotes Leishmania virulence. J. Biol. Chem. 2017, 292, 12324–12338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romao, P.R.; Tovar, J.; Fonseca, S.G.; Moraes, R.H.; Cruz, A.K.; Hothersall, J.S.; Noronha-Dutra, A.A.; Ferreira, S.H.; Cunha, F.Q. Glutathione and the redox control system trypanothione/trypanothione reductase are involved in the protection of Leishmania spp. against nitrosothiol-induced cytotoxicity. Braz. J. Med. Biol. Res. 2006, 39, 355–363. [Google Scholar] [CrossRef]
- Ribeiro, R.R.; Michalick, M.S.M.; da Silva, M.E.; Dos Santos, C.C.P.; Frézard, F.J.G.; da Silva, S.M. Canine Leishmaniasis: An Overview of the Current Status and Strategies for Control. Biomed. Res. Int. 2018, 3296893. [Google Scholar] [CrossRef]
- Dantas-Torres, F.; Miró, G.; Baneth, G.; Bourdeau, P.; Breitschwerdt, E.; Capelli, G.; Cardoso, L.; Day, M.J.; Dobler, G.; Ferrer, L.; et al. Canine Leishmaniasis control in the context of one health. Emerg. Infect. Dis. 2019, 25, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Hotez, P.J. Neglected tropical diseases in the ebola-affected countries of West Africa. PLoS Negl. Trop. Dis. 2015, 9, e0003671. [Google Scholar] [CrossRef]
- Molyneux, D.H.; Hotez, P.J.; Fenwick, A. “Rapid-impact interventions”: How a policy of integrated control for Africa’s neglected tropical diseases could benefit the poor. PLoS Med. 2005, 2, e336. [Google Scholar] [CrossRef] [Green Version]
- Kirigia, J.M.; Mburugu, G.N. The monetary value of human lives lost due to neglected tropical diseases in Africa. Infect. Dis. Poverty 2017, 6, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebremichael Tedla, D.; Bariagabr, F.H.; Abreha, H.H. Incidence and trends of Leishmaniasis and its risk factors in humera, western tigray. J. Parasitol. Res. 2018, 2018, 8463097. [Google Scholar] [CrossRef] [Green Version]
- WHO. Disease watch focus: Leishmaniasis. TDR Nat. Rev. Microbiol. 2004, 2, 6926–6993. [Google Scholar]
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Cota, G.F.; de Sousa, M.R.; Rabello, A. Predictors of visceral leishmaniasis relapse in HIV-infected patients: A systematic review. PLoS Negl. Trop. Dis. 2011, 5, e1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, J.N.; Lockwood, D.N. Clinical aspects of visceral leishmaniasis in HIV infection. Curr. Opin. Infect. Dis. 2013, 26, 1–9. [Google Scholar] [CrossRef]
- Lindoso, J.A.L.; Moreira, C.H.V.; Cunha, M.A.; Queiroz, I.T. Visceral leishmaniasis and HIV coinfection: Current perspectives. HIV AIDS (Auckl.) 2018, 10, 1932–2001. [Google Scholar] [CrossRef] [Green Version]
- WHO. Progress Report: Global HIV/AIDS Response; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Bankoti, R.; Stager, S. Differential regulation of the immune response in the spleen and liver of mice infected with Leishmania donovani. J. Trop. Med. 2012, 2012, 639304. [Google Scholar] [CrossRef] [Green Version]
- Egui, A.; Ledesma, D.; Perez-Anton, E.; Montoya, A.; Gomez, I.; Robledo, S.M.; Infante, J.J.; Vélez, I.D.; López, M.C.; Thomas, M.C. Phenotypic and functional profiles of antigen-specific CD4(+) and CD8(+) T cells associated with infection control in patients with cutaneous Leishmaniasis. Front. Cell. Infect. Microbiol. 2018, 8, 393. [Google Scholar] [CrossRef]
- Hurdayal, R.; Ndlovu, H.H.; Revaz-Breton, M.; Parihar, S.P.; Nono, J.K.; Govender, M.; Brombacher, F. IL-4-producing B cells regulate T helper cell dichotomy in type 1- and type 2-controlled diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E8430–E8439. [Google Scholar] [CrossRef] [Green Version]
- Masic, A.; Hurdayal, R.; Nieuwenhuizen, N.E.; Brombacher, F.; Moll, H. Dendritic cell-mediated vaccination relies on interleukin-4 receptor signaling to avoid tissue damage after Leishmania major infection of BALB/c mice. PLoS Negl. Trop. Dis. 2012, 6, e1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarlane, E.; Mokgethi, T.; Kaye, P.M.; Hurdayal, R.; Brombacher, F.; Alexander, J.; Carter, K.C. IL-4 Mediated Resistance of BALB/c Mice to Visceral Leishmaniasis is independent of IL-4Ralpha signaling via T cells. Front. Immunol. 2019, 10, 1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descatoire, M.; Hurrell, B.P.; Govender, M.; Passelli, K.; Martinez-Salazar, B.; Hurdayal, R.; Brombacher, F.; Guler, R.; Tacchini-Cottier, F. IL-4Ralpha signaling in keratinocytes and early IL-4 production are dispensable for generating a curative T Helper 1 response in Leishmania major-infected C57BL/6 Mice. Front. Immunol. 2017, 8, 1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangert, M.; Molyneux, D.H.; Lindsay, S.W.; Fitzpatrick, C.; Engels, D. The cross-cutting contribution of the end of neglected tropical diseases to the sustainable development goals. Infect. Dis. Poverty 2017, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; Lopez-Velez, R.; Garcia-Hernandez, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in Leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral Leishmaniasis: Present and future developments. Parasitology 2018, 145, 4814–4889. [Google Scholar] [CrossRef]
- Haldar, A.K.; Sen, P.; Roy, S. Use of antimony in the treatment of Leishmaniasis: Current status and future directions. Mol. Biol. Int. 2011, 2011, 571242. [Google Scholar] [CrossRef] [Green Version]
- Rojas, R.; Valderrama, L.; Valderrama, M.; Varona, M.X.; Ouellette, M.; Saravia, N.G. Resistance to antimony and treatment failure in human Leishmania (Viannia) infection. J. Infect. Dis. 2006, 193, 1375–1383. [Google Scholar] [CrossRef] [Green Version]
- Chulay, J.D.; Fleckenstein, L.; Smith, D.H. Pharmacokinetics of antimony during treatment of visceral Leishmaniasis with sodium stibogluconate or meglumine antimoniate. Trans. R. Soc. Trop. Med. Hyg. 1988, 82, 697–702. [Google Scholar] [CrossRef]
- Frezard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent antimonials: New perspectives for old drugs. Molecules 2009, 14, 2317–2336. [Google Scholar] [CrossRef] [Green Version]
- Rijal, S.; Chappuis, F.; Singh, R.; Boelaert, M.; Loutan, L.; Koirala, S. Sodium stibogluconate cardiotoxicity and safety of generics. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 597–598. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; More, D.K.; Singh, M.K.; Singh, V.P.; Sharma, S.; Makharia, A.; Kumar, P.C.; Murray, H.W. Failure of pentavalent antimony in visceral Leishmaniasis in India: Report from the center of the Indian epidemic. Clin. Infect. Dis. 2000, 31, 1104–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesur, S.; Bahar, K.; Erekul, S. Death from cumulative sodium stibogluconate toxicity on Kala-Azar. Clin. Microbiol. Infect. 2002, 8, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikrant, S.; Gupta, D.; Kaushal, S.S. Sodium stibogluconate-associated acute interstitial nephritis in a patient treated for visceral Leishmaniasis. Saudi. J. Kidney Dis. Transpl. 2015, 26, 757–760. [Google Scholar] [CrossRef]
- Goodwin, L.G.; Page, J.E. A study of the excretion of organic antimonials using a polarographic procedure. Biochem. J. 1943, 37, 1982–2009. [Google Scholar] [CrossRef] [Green Version]
- Shaked-Mishan, P.; Ulrich, N.; Ephros, M.; Zilberstein, D. Novel Intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 2001, 276, 3971–3976. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; McMaster, W.R. Leishmania major HEXBP deletion mutants generated by double targeted gene replacement. Mol. Biochem. Parasitol. 1994, 63, 231–242. [Google Scholar] [CrossRef]
- Krauth-Siegel, R.L.; Comini, M.A. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim. Biophys. Acta 2008, 1780, 1236–1248. [Google Scholar] [CrossRef]
- Demicheli, C.; Frezard, F.; Lecouvey, M.; Garnier-Suillerot, A. Antimony(V) complex formation with adenine nucleosides in aqueous solution. Biochim. Biophys. Acta 2002, 1570, 192–198. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, A. Recent developments and future prospects in the treatment of visceral Leishmaniasis. Ther. Adv. Infect. Dis. 2016, 3, 981–1009. [Google Scholar] [CrossRef]
- Alexander, J.; Carter, K.C.; Al-Fasi, N.; Satoskar, A.; Brombacher, F. Endogenous IL-4 is necessary for effective drug therapy against visceral Leishmaniasis. Eur. J. Immunol. 2000, 30, 2935–2943. [Google Scholar] [CrossRef]
- Murray, H.W. Clinical and experimental advances in treatment of visceral Leishmaniasis. Antimicrob. Agents Chemother. 2001, 45, 2185–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, H.W.; Delph-Etienne, S. Roles of endogenous gamma interferon and macrophage microbicidal mechanisms in host response to chemotherapy in experimental visceral Leishmaniasis. Infect. Immun. 2000, 68, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, H.W.; Hariprashad, J.; Fichtl, R.E. Treatment of experimental visceral Leishmaniasis in a T-cell-deficient host: Response to amphotericin B and pentamidine. Antimicrob. Agents Chemother. 1993, 37, 1504–1505. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.W.; Montelibano, C.; Peterson, R.; Sypek, J.P. Interleukin-12 regulates the response to chemotherapy in experimental visceral Leishmaniasis. J. Infect. Dis. 2000, 182, 1497–1502. [Google Scholar] [CrossRef]
- Mookerjee Basu, J.; Mookerjee, A.; Sen, P.; Bhaumik, S.; Sen, P.; Banerjee, S.; Naskar, K.; Choudhur, S.K.; Saha, B.; Raha, J.P.; et al. Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob Agents Chemother. 2006, 50, 1788–1797. [Google Scholar] [CrossRef] [Green Version]
- Carter, K.C.; Baillie, A.J.; Alexander, J.; Dolan, T.F. The therapeutic effect of sodium stibogluconate in BALB/c mice infected with Leishmania donovani is organ-dependent. J. Pharm. Pharmacol. 1988, 40, 370–373. [Google Scholar] [CrossRef]
- Collins, M.; Carter, K.C.; Baillie, A.J. Visceral leishmaniasis in the BALB/c mouse: Antimony tissue disposition and parasite suppression after the administration of free stibogluconate. Ann. Trop. Med. Parasitol. 1992, 86, 35–40. [Google Scholar] [CrossRef]
- Murray, H.W. Tissue granuloma structure-function in experimental visceral Leishmaniasis. Int. J. Exp. Pathol. 2001, 82, 249–267. [Google Scholar] [CrossRef]
- Ashutosh; Sundar, S.; Goyal, N. Molecular mechanisms of antimony resistance in Leishmania. J. Med. Microbiol. 2007, 56, 143–153. [Google Scholar] [CrossRef]
- Brochu, C.; Wang, J.; Roy, G.; Messier, N.; Wang, X.Y.; Saravia, N.G.; Ouellette, M. Antimony uptake systems in the protozoan parasite Leishmania and accumulation differences in antimony-resistant parasites. Antimicrob. Agents Chemother. 2003, 47, 3073–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, S.; Bharati, K.; Shaha, C.; Mukhopadhyay, C.K. Zinc depletion promotes apoptosis-like death in drug-sensitive and antimony-resistance Leishmania donovani. Sci. Rep. 2017, 7, 10488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, S.; Singh, B. Understanding Leishmania parasites through proteomics and implications for the clinic. Expert Rev Proteomics. 2018, 15, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Yardley, V. Chemotherapy of Leishmaniasis. Curr. Pharm. Des. 2002, 8, 319–342. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Bigot, S.; Padmanabhan, P.K.; Mukherjee, A.; Coelho, A.; Leprohon, P.; Papadopoulou, B.; Ouellette, M. New insights in the mode of action of anti-leishmanial drugs by using chemical mutagenesis screens coupled to next-generation sequencing. Microb. Cell 2020, 7, 596–601. [Google Scholar] [CrossRef]
- Decuypere, S.; Rijal, S.; Yardley, V.; De Doncker, S.; Laurent, T.; Khanal, B.; Chappuis, F.; Dujardin, J.C. Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob. Agents Chemother. 2005, 49, 4616–4621. [Google Scholar] [CrossRef] [Green Version]
- Patino, L.H.; Imamura, H.; Cruz-Saavedra, L.; Pavia, P.; Muskus, C.; Mendez, C.; Dujardin, J.C.; Ramírez, J.D. Major changes in chromosomal somy, gene expression and gene dosage driven by Sb(III) in Leishmania braziliensis and Leishmania panamensis. Sci. Rep. 2019, 9, 9485. [Google Scholar] [CrossRef]
- Perea, A.; Manzano, J.I.; Castanys, S.; Gamarro, F. The LABCG2 transporter from the protozoan parasite Leishmania is involved in antimony resistance. Antimicrob. Agents Chemother. 2016, 60, 3489–3496. [Google Scholar] [CrossRef] [Green Version]
- Marquis, N.; Gourbal, B.; Rosen, B.P.; Mukhopadhyay, R.; Ouellette, M. Modulation in aquaglyceroporin AQP1 gene transcript levels in drug-resistant Leishmania. Mol. Microbiol. 2005, 57, 1690–1699. [Google Scholar] [CrossRef]
- Gallis, H.A.; Drew, R.H.; Pickard, W.W. Amphotericin B: 30 years of clinical experience. Rev. Infect. Dis. 1990, 12, 308–329. [Google Scholar] [CrossRef]
- Sundar, S.; Agrawal, G.; Rai, M.; Makharia, M.K.; Murray, H.W. Treatment of Indian visceral Leishmaniasis with single or daily infusions of low dose liposomal amphotericin B: Randomised trial. BMJ 2001, 323, 419–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, S.; Chakravarty, J.; Rai, V.K.; Agrawal, N.; Singh, S.P.; Chauhan, V.; Murray, H.W. Amphotericin B treatment for Indian visceral leishmaniasis: Response to 15 daily versus alternate-day infusions. Clin. Infect. Dis. 2007, 45, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.P.; Singh, R.K.; Hassan, S.M.; Kumar, R.; Narain, S.; Kumar, A. Amphotericin B deoxycholate treatment of visceral leishmaniasis with newer modes of administration and precautions: A study of 938 cases. Trans. R. Soc. Trop. Med. Hyg. 1999, 93, 319–323. [Google Scholar] [CrossRef]
- Deray, G. Amphotericin B nephrotoxicity. J. Antimicrob. Chemother. 2002, 49 (Suppl. 1), 374–381. [Google Scholar] [CrossRef]
- Fernandez-Garcia, R.; de Pablo, E.; Ballesteros, M.P.; Serrano, D.R. Unmet clinical needs in the treatment of systemic fungal infections: The role of Amphotericin B and drug targeting. Int. J. Pharm. 2017, 525, 1391–1448. [Google Scholar] [CrossRef]
- Dietze, R.; Fagundes, S.M.; Brito, E.F.; Milan, E.P.; Feitosa, T.F.; Suassuna, F.A.; Fonschiffrey, G.; Ksionski, G.; Dember, J. Treatment of kala-azar in Brazil with Amphocil (amphotericin B cholesterol dispersion) for 5 days. Trans. R. Soc. Trop. Med. Hyg. 1995, 89, 309–311. [Google Scholar] [CrossRef]
- Syriopoulou, V.; Daikos, G.L.; Theodoridou, M.; Pavlopoulou, I.; Manolaki, A.G.; Sereti, E.; Karamboula, A.; Papathanasiou, D.; Krikos, X.; Saroglou, G. Two doses of a lipid formulation of amphotericin B for the treatment of Mediterranean visceral Leishmaniasis. Clin. Infect. Dis. 2003, 36, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Groll, A.H.; Rijnders, B.J.A.; Walsh, T.J.; Adler-Moore, J.; Lewis, R.E.; Bruggemann, R.J.M. Clinical pharmacokinetics, pharmacodynamics, safety and efficacy of liposomal Amphotericin B. Clin. Infect. Dis. 2019, 68, S260–S274. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J.; Agarwal, D.; Rai, M.; Murray, H.W. Single-dose liposomal Amphotericin B for visceral Leishmaniasis in India. N. Engl. J. Med. 2010, 362, 504–512. [Google Scholar] [CrossRef]
- Sundar, S.; Mehta, H.; Suresh, A.V.; Singh, S.P.; Rai, M.; Murray, H.W. Amphotericin B treatment for Indian visceral Leishmaniasis: Conventional versus lipid formulations. Clin. Infect. Dis. 2004, 38, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Guery, R.; Henry, B.; Martin-Blondel, G.; Rouzaud, C.; Cordoliani, F.; Harms, G.; Gangneux, J.P.; Foulet, F.; Bourrat, E.; Baccard, M.; et al. Liposomal amphotericin B in travelers with cutaneous and muco-cutaneous leishmaniasis: Not a panacea. PLoS Negl. Trop. Dis. 2017, 11, e0006094. [Google Scholar] [CrossRef]
- Mosimann, V.; Neumayr, A.; Paris, D.H.; Blum, J. Liposomal Amphotericin B treatment of Old World cutaneous and mucosal Leishmaniasis: A literature review. Acta Trop. 2018, 182, 2462–2550. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, G.; Zapor, M.; Ressner, R.; Fraser, S.; Hartzell, J.; Pierson, J.; Weintrob, A.; Magill, A. Lipsosomal amphotericin B for treatment of cutaneous Leishmaniasis. Am. J. Trop. Med. Hyg. 2010, 83, 1028–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichenberger, A.; Buechi, A.E.; Neumayr, A.; Hatz, C.; Rauch, A.; Huguenot, M.; Diamantis-Karamitopoulou, E.; Staehelin, C. A severe case of visceral leishmaniasis and liposomal amphotericin B treatment failure in an immunosuppressed patient 15 years after exposure. BMC Infect. Dis. 2017, 17, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Control of the Leishmaniases. Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Mesa-Arango, A.C.; Scorzoni, L.; Zaragoza, O. It only takes one to do many jobs: Amphotericin B as antifungal and immunomodulatory drug. Front. Microbiol. 2012, 3, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, J.; Saxena, A.; Singh, S. Chemotherapy of Leishmaniasis: Past, present and future. Curr. Med. Chem. 2007, 14, 1153–1169. [Google Scholar] [CrossRef]
- Ramos, H.; Saint-Pierre-Chazalet, M.; Bolard, J.; Cohen, B.E. Effect of ketoconazole on lethal action of amphotericin B on Leishmania mexicana promastigotes. Antimicrob. Agents Chemother. 1994, 38, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.W.; Brooks, E.B.; DeVecchio, J.L.; Heinzel, F.P. Immunoenhancement combined with amphotericin B as treatment for experimental visceral Leishmaniasis. Antimicrob. Agents Chemother. 2003, 47, 2513–2517. [Google Scholar] [CrossRef] [Green Version]
- van Griensven, J.; Carrillo, E.; Lopez-Velez, R.; Lynen, L.; Moreno, J. Leishmaniasis in immunosuppressed individuals. Clin. Microbiol. Infect. 2014, 20, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Purkait, B.; Kumar, A.; Nandi, N.; Sardar, A.H.; Das, S.; Kumar, S.; Pandey, K.; Ravidas, V.; Kumar, M.; De, T.; et al. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob. Agents Chemother. 2012, 56, 1031–1041. [Google Scholar] [CrossRef] [Green Version]
- Mwenechanya, R.; Kovarova, J.; Dickens, N.J.; Mudaliar, M.; Herzyk, P.; Vincent, I.M.; Weidt, S.K.; Burgess, K.E.; Burchmore, R.J.; Pountain, A.W.; et al. Sterol 14alpha-demethylase mutation leads to amphotericin B resistance in Leishmania mexicana. PLoS Negl. Trop. Dis. 2017, 11, e0005649. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.D.; Chen, L.L.; Tai, P.C. Misread protein creates membrane channels: An essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. USA 1986, 83, 6164–6168. [Google Scholar]
- Gardner, T.B.; Hill, D.R. Treatment of giardiasis. Clin. Microbiol. Rev. 2001, 14, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, T.; Koga, M.; Shimizu, S.; Miura, T.; Maruyama, H.; Kimura, M. Efficacy and safety of paromomycin for treating amebiasis in Japan. Parasitol. Int. 2013, 62, 4975–5001. [Google Scholar] [CrossRef] [PubMed]
- Antinori, S.; Schifanella, L.; Corbellino, M. Leishmaniasis: New insights from an old and neglected disease. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Ben Salah, A.; Ben Messaoud, N.; Guedri, E.; Zaatour, A.; Ben Alaya, N.; Bettaieb, J.; Gharbi, A.; Belhadj Hamida, N.; Boukthir, A.; Chlif, S.; et al. Topical paromomycin with or without gentamicin for cutaneous Leishmaniasis. N. Engl. J. Med. 2013, 368, 524–532. [Google Scholar] [PubMed] [Green Version]
- Sundar, S.; Chatterjee, M. Visceral Leishmaniasis-current therapeutic modalities. Indian J. Med. Res. 2006, 123, 345–352. [Google Scholar]
- Davidson, R.N.; den Boer, M.; Ritmeijer, K. Paromomycin. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 653–660. [Google Scholar] [CrossRef]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Sinha, P.K.; Bhattacharya, S.K. Injectable paromomycin for Visceral leishmaniasis in India. N. Engl. J. Med. 2007, 356, 2571–2581. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Chung, H.J.; Bleys, J.; Ghohestani, R.F. Is paromomycin an effective and safe treatment against cutaneous Leishmaniasis? A meta-analysis of 14 randomized controlled trials. PLoS Negl. Trop. Dis. 2009, 3, e381. [Google Scholar] [CrossRef] [Green Version]
- Moradzadeh, R.; Golmohammadi, P.; Ashraf, H.; Nadrian, H.; Fakoorziba, M.R. Effectiveness of Paromomycin on Cutaneous Leishmaniasis in Iran: A Systematic Review and Meta-Analysis. Iran. J. Med. Sci. 2019, 44, 1851–1895. [Google Scholar]
- Maarouf, M.; de Kouchkovsky, Y.; Brown, S.; Petit, P.X.; Robert-Gero, M. In vivo interference of paromomycin with mitochondrial activity of Leishmania. Exp. Cell Res. 1997, 232, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, M.; Lawrence, F.; Brown, S.; Robert-Gero, M. Biochemical alterations in paromomycin-treated Leishmania donovani promastigotes. Parasitol. Res. 1997, 83, 1982–2002. [Google Scholar] [CrossRef] [PubMed]
- Shalev-Benami, M.; Zhang, Y.; Rozenberg, H.; Nobe, Y.; Taoka, M.; Matzov, D.; Zimmerman, E.; Bashan, A.; Isobe, T.; Jaffe, C.L.; et al. Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat. Commun. 2017, 8, 1589. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, S.; Inocencio da Luz, R.A.; Bhandari, V.; Kuypers, K.; Shaw, C.D.; Lonchamp, J.; Salotra, P.; Carter, K.; Sundar, S.; Rijal, S.; et al. Experimental induction of paromomycin resistance in antimony-resistant strains of L. donovani: Outcome dependent on in vitro selection protocol. PLoS Negl. Trop. Dis. 2012, 6, e1664. [Google Scholar]
- Hendrickx, S.; Mondelaers, A.; Eberhardt, E.; Delputte, P.; Cos, P.; Maes, L. In vivo selection of paromomycin and Miltefosine Resistance in Leishmania donovani and L. infantum in a Syrian Hamster Model. Antimicrob. Agents Chemother. 2015, 59, 4714–4718. [Google Scholar] [CrossRef] [Green Version]
- Shaw, C.D.; Imamura, H.; Downing, T.; Blackburn, G.; Westrop, G.D.; Cotton, J.A.; Berriman, M.; Sanders, M.; Rijal, S.; Coombs, G.H.; et al. Genomic and metabolomic polymorphism among experimentally selected paromomycin-resistant Leishmania donovani strains. Antimicrob. Agents Chemother. 2019, 64, e00904–e00919. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Gupta, L.B.; Makharia, M.K.; Singh, M.K.; Voss, A.; Rosenkaimer, F.; Engel, J.; Murray, H.W. Oral treatment of visceral leishmaniasis with miltefosine. Ann. Trop. Med. Parasitol. 1999, 93, 589–597. [Google Scholar] [CrossRef]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral miltefosine for Indian visceral Leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.K.; Sinha, P.K.; Sundar, S.; Thakur, C.P.; Jha, T.K.; Pandey, K.; Das, V.R.; Kumar, N.; Lal, C.; Verma, N.; et al. Phase 4 trial of miltefosine for the treatment of Indian visceral Leishmaniasis. J. Infect. Dis. 2007, 196, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Wasunna, M.; Njenga, S.; Balasegaram, M.; Alexander, N.; Omollo, R.; Edwards, T.; Dorlo, T.P.; Musa, B.; Ali, M.H.S.; Elamin, M.Y.; et al. Efficacy and safety of AmBisome in combination with Sodium Stibogluconate or Miltefosine and Miltefosine Monotherapy for African Visceral Leishmaniasis: Phase II Randomized Trial. PLoS Negl. Trop. Dis. 2016, 10, e0004880. [Google Scholar] [CrossRef] [PubMed]
- Ritmeijer, K.; Dejenie, A.; Assefa, Y.; Hundie, T.B.; Mesure, J.; Boots, G.; den Boer, M. A comparison of Miltefosine and sodium stibogluconate for treatment of visceral Leishmaniasis in an Ethiopian population with high prevalence of HIV infection. Clin. Infect. Dis. 2006, 43, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, J.; Arana, B.A.; Toledo, J.; Rizzo, N.; Vega, J.C.; Diaz, A.; Luz, M.; Gutierrez, P.; Arboleda, M.; Berman, J.D.; et al. Miltefosine for new world cutaneous Leishmaniasis. Clin. Infect. Dis. 2004, 38, 1266–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, P.R.; Ampuero, J.; Guimaraes, L.H.; Villasboas, L.; Rocha, A.T.; Schriefer, A.; Sousa, R.S.; Talhari, A.; Penna, G.; Carvalho, E.M. Miltefosine in the treatment of cutaneous leishmaniasis caused by Leishmania braziliensis in Brazil: A randomized and controlled trial. PLoS Negl. Trop. Dis. 2010, 4, e912. [Google Scholar] [CrossRef] [Green Version]
- Calvopina, M.; Gomez, E.A.; Sindermann, H.; Cooper, P.J.; Hashiguchi, Y. Relapse of new world diffuse cutaneous Leishmaniasis caused by Leishmania (Leishmania) mexicana after miltefosine treatment. Am. J. Trop. Med. Hyg. 2006, 75, 1074–1077. [Google Scholar] [CrossRef] [Green Version]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of Leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- Pinto-Martinez, A.K.; Rodriguez-Duran, J.; Serrano-Martin, X.; Hernandez-Rodriguez, V.; Benaim, G. Mechanism of action of miltefosine on Leishmania donovani involves the impairment of acidocalcisome function and the activation of the sphingosine-dependent plasma membrane Ca(2+) channel. Antimicrob. Agents Chemother. 2018, 62, e01614–e01617. [Google Scholar] [CrossRef] [Green Version]
- Palic, S.; Bhairosing, P.; Beijnen, J.H.; Dorlo, T.P.C. Systematic review of host-mediated activity of Miltefosine in Leishmaniasis through immunomodulation. Antimicrob. Agents Chemother. 2019, 63, e02507–e02518. [Google Scholar] [CrossRef] [Green Version]
- Ponte, C.B.; Alves, E.A.; Sampaio, R.N.; Urdapilleta, A.A.; Kuckelhaus Cdos, S.; Muniz-Junqueira, M.I.; Kuckelhaus, S.A. Miltefosine enhances phagocytosis but decreases nitric oxide production by peritoneal macrophages of C57BL/6 mice. Int. Immunopharmacol. 2012, 13, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Wadhone, P.; Maiti, M.; Agarwal, R.; Kamat, V.; Martin, S.; Saha, B. Miltefosine promotes IFN-gamma-dominated anti-leishmanial immune response. J. Immunol. 2009, 182, 7146–7154. [Google Scholar] [CrossRef] [Green Version]
- Perez-Victoria, F.J.; Sanchez-Canete, M.P.; Castanys, S.; Gamarro, F. Phospholipid translocation and miltefosine potency require both L. donovani miltefosine transporter and the new protein LdRos3 in Leishmania parasites. J. Biol. Chem. 2006, 281, 23766–23775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, C.D.; Lonchamp, J.; Downing, T.; Imamura, H.; Freeman, T.M.; Cotton, J.A.; Sanders, M.; Blackburn, G.; Dujardin, J.C.; Rijal, S.; et al. In vitro selection of miltefosine resistance in promastigotes of Leishmania donovani from Nepal: Genomic and metabolomic characterization. Mol. Microbiol. 2016, 99, 1134–1148. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Singh, A.; Rai, M.; Prajapati, V.K.; Singh, A.K.; Ostyn, B.; Boelaert, M.; Dujardin, J.C.; Chakravarty, J. Efficacy of miltefosine in the treatment of visceral Leishmaniasis in India after a decade of use. Clin. Infect. Dis. 2012, 55, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Mishra, J.; Gupta, A.K.; Singh, A.; Shankar, P.; Singh, S. Laboratory confirmed miltefosine resistant cases of visceral leishmaniasis from India. Parasites Vectors 2017, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Varikuti, S.; Jha, B.K.; Volpedo, G.; Ryan, N.M.; Halsey, G.; Hamza, O.M.; McGwire, B.S.; Satoskar, A.R. Host-Directed drug therapies for neglected tropical diseases caused by protozoan parasites. Front. Microbiol. 2018, 9, 2655. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-directed therapies for infectious diseases: Current status, recent progress, and future prospects. Lancet Infect. Dis. 2016, 16, e476–e483. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.W.; Miralles, G.D.; Stoeckle, M.Y.; McDermott, D.F. Role and effect of IL-2 in experimental visceral leishmaniasis. J. Immunol. 1993, 151, 929–938. [Google Scholar]
- Murray, H.W.; Hariprashad, J. Interleukin 12 is effective treatment for an established systemic intracellular infection: Experimental visceral Leishmaniasis. J. Exp. Med. 1995, 181, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Murray, H.W. Effect of treatment with interferon-gamma alone in visceral Leishmaniasis. J. Infect. Dis. 1995, 172, 1627–1629. [Google Scholar] [PubMed]
- Badaro, R.; Falcoff, E.; Badaro, F.S.; Carvalho, E.M.; Pedral-Sampaio, D.; Barral, A.; Carvalho, J.S.; Barral-Netto, M.; Brandely, M.; Silva, L.; et al. Treatment of visceral Leishmaniasis with pentavalent antimony and interferon gamma. N. Engl. J. Med. 1990, 322, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Cook, A.M.; McDonnell, A.M.; Millward, M.J.; Creaney, J.; Francis, R.J.; Hasani, A.; Segal, A.; Musk, A.W.; Turlach, B.A.; et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann. Oncol. 2015, 26, 2483–2490. [Google Scholar] [CrossRef]
- Chan, D.V.; Gibson, H.M.; Aufiero, B.M.; Wilson, A.J.; Hafner, M.S.; Mi, Q.S.; Wong, H.K. Differential CTLA-4 expression in human CD4+ versus CD8+ T cells is associated with increased NFAT1 and inhibition of CD4+ proliferation. Genes Immun. 2014, 15, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.L.; Cotterell, S.E.; Gorak, P.M.; Engwerda, C.R.; Kaye, P.M. Blockade of CTLA-4 enhances host resistance to the intracellular pathogen, Leishmania donovani. J. Immunol. 1998, 161, 4153–4160. [Google Scholar]
- Murray, H.W.; Lu, C.M.; Brooks, E.B.; Fichtl, R.E.; DeVecchio, J.L.; Heinzel, F.P. Modulation of T-cell costimulation as immunotherapy or immunochemotherapy in experimental visceral Leishmaniasis. Infect. Immun. 2003, 71, 6453–6462. [Google Scholar] [CrossRef] [Green Version]
- Zubairi, S.; Sanos, S.L.; Hill, S.; Kaye, P.M. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur. J. Immunol. 2004, 34, 1433–1440. [Google Scholar] [CrossRef]
- Biedermann, T.; Zimmermann, S.; Himmelrich, H.; Gumy, A.; Egeter, O.; Sakrauski, A.K.; Seegmuller, I.; Voigt, H.; Launois, P.; Levine, A.D.; et al. IL-4 instructs TH1 responses and resistance to Leishmania major in susceptible BALB/c mice. Nat. Immunol. 2001, 2, 1054–1060. [Google Scholar] [CrossRef]
- Hurdayal, R.; Nieuwenhuizen, N.E.; Revaz-Breton, M.; Smith, L.; Hoving, J.C.; Parihar, S.P.; Reizis, B.; Brombacher, F. Deletion of IL-4 receptor alpha on dendritic cells renders BALB/c mice hypersusceptible to Leishmania major infection. PLoS Pathog. 2013, 9, e1003699. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Li, W.; Kaplan, M.H.; Chang, C.H. Interleukin (IL)-4 inhibits IL-10 to promote IL-12 production by dendritic cells. J. Exp. Med. 2005, 201, 1899–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurdayal, R.; Brombacher, F. Interleukin-4 receptor alpha: From innate to adaptive immunity in murine models of cutaneous Leishmaniasis. Front. Immunol. 2017, 8, 1354. [Google Scholar] [CrossRef]
- Hurdayal, R.; Brombacher, F. The role of IL-4 and IL-13 in cutaneous Leishmaniasis. Immunol. Lett. 2014, 161, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Pal, C.; Ray, M.; Maitra, S.; Mandal, L.; Bandyopadhyay, S. Dendritic cell-based immunotherapy combined with antimony-based chemotherapy cures established murine visceral Leishmaniasis. J. Immunol. 2003, 170, 5625–5629. [Google Scholar] [PubMed]
- Dayakar, A.; Chandrasekaran, S.; Kuchipudi, S.V.; Kalangi, S.K. Cytokines: Key determinants of resistance or disease progression in visceral Leishmaniasis: Opportunities for novel diagnostics and immunotherapy. Front. Immunol. 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, H.W. Interleukin 10 receptor blockade--pentavalent antimony treatment in experimental visceral Leishmaniasis. Acta Trop. 2005, 93, 2953–3001. [Google Scholar] [CrossRef]
- Murray, H.W.; Lu, C.M.; Mauze, S.; Freeman, S.; Moreira, A.L.; Kaplan, G.; Coffman, R.L. Interleukin-10 (IL-10) in experimental visceral leishmaniasis and IL-10 receptor blockade as immunotherapy. Infect. Immun. 2002, 70, 6284–6293. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.W.; Moreira, A.L.; Lu, C.M.; DeVecchio, J.L.; Matsuhashi, M.; Ma, X.; Heinzel, F.P. Determinants of response to interleukin-10 receptor blockade immunotherapy in experimental visceral Leishmaniasis. J. Infect. Dis. 2003, 188, 458–464. [Google Scholar] [PubMed]
- Rub, A.; Arish, M.; Husain, S.A.; Ahmed, N.; Akhter, Y. Host-lipidome as a potential target of protozoan parasites. Microbes Infect. 2013, 15, 649–660. [Google Scholar] [CrossRef]
- Parihar, S.P.; Guler, R.; Khutlang, R.; Lang, D.M.; Hurdayal, R.; Mhlanga, M.M.; Suzuki, H.; Marais, A.D.; Brombacher, F. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 2014, 209, 754–763. [Google Scholar]
- Bouazizi-Ben Messaoud, H.; Guichard, M.; Lawton, P.; Delton, I.; Azzouz-Maache, S. Changes in lipid and fatty acid composition during intramacrophagic transformation of Leishmania donovani complex promastigotes into amastigotes. Lipids 2017, 52, 4334–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.C.; Yang, P.W.; Yang, S.F.; Hsieh, K.P.; Tseng, S.P.; Lin, Y.C. Topical simvastatin promotes healing of Staphylococcus aureus-contaminated cutaneous wounds. Int. Wound J. 2016, 13, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; De, M.; Ali, N. Combination therapy with paromomycin-associated stearylamine-bearing liposomes cures experimental visceral Leishmaniasis through Th1-biased immunomodulation. Antimicrob. Agents Chemother. 2011, 55, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.; Ravindran, R.; Ali, N. Combination therapy using sodium antimony gluconate in stearylamine-bearing liposomes against established and chronic Leishmania donovani infection in BALB/c Mice. Antimicrob. Agents Chemother. 2004, 48, 3591–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speirs, K.; Caamano, J.; Goldschmidt, M.H.; Hunter, C.A.; Scott, P. NF-kappa B2 is required for optimal CD40-induced IL-12 production but dispensable for Th1 cell differentiation. J. Immunol. 2002, 168, 4406–4413. [Google Scholar] [CrossRef] [Green Version]
- Ben-Othman, R.; Dellagi, K.; Guizani-Tabbane, L. Leishmania major parasites induced macrophage tolerance: Implication of MAPK and NF-kappaB pathways. Mol. Immunol. 2009, 46, 3438–3444. [Google Scholar] [CrossRef]
- Luthuli, S.; Wu, S.; Cheng, Y.; Zheng, X.; Wu, M.; Tong, H. Therapeutic effects of fucoidan: A review on recent studies. Mar. Drugs 2019, 17, 487. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Kar, S.; Basu Ball, W.; Ghosh, K.; Das, P.K. The curative effect of fucoidan on visceral leishmaniasis is mediated by activation of MAP kinases through specific protein kinase C isoforms. Cell. Mol. Immunol. 2014, 11, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Ukil, A.; Das, P.K. Signaling events leading to the curative effect of cystatin on experimental visceral Leishmaniasis: Involvement of ERK1/2, NF-kappaB and JAK/STAT pathways. Eur. J. Immunol. 2009, 39, 741–751. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 163–172. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Leishmania Spp | Vector | OW/NW | Location | Type of Disease |
|---|---|---|---|---|
| Leishmania major | Phlebotomus papatasi P. ansari P. caucasicus P. bergeroti P. sergenti | OW | Middle East, North Africa, Asia | CL |
| Leishmania donovani | P. argentipes P. martini P. chinensis P. orientalis P. alexandri P. celiae | OW | East Africa, India subcontinent | VL |
| Leishmania infantum | P. alexandri P. ariasi P. langeroni P. longicuspis Lutzomyia migonei L. longipalpis L. cortelezzii | OW | Central and South America, Mediterranean regions, Asia | VL |
| Leishmania siamensis | Sergentomyia (Neophlebotomus) gemmea | NW | Thailand, USA, Central and Western Europe | DCL/VL |
| Leishmania braziliensis | L. longipalpis L. ayrozai L. lichyi Warileya rotundipennis L. shawi L. whitmani L. (Pintomyia) fischeri L. wellcomei | NW | South America | MCL |
| Leishmania mexicana | L. gomezi L. trapidoi L. anthophora L. ovallesi L. diabolica | NW | North and South America | CL |
| Leishmania amazonensis | L. evansi L. diabolica L. longipalpis L. (Nyssomyia) flaviscutellata | NW | Amanzonas | CL/MCL |
| Leishmania venezuelensis | L. olmeca L. lichyi L. rangeliana | NW | Western Venezuela | CL |
| Leishmania aethiopica | P. longipes P. sergenti | OW | East Africa | CL/MCL |
| Leishmania tropica | P. guggisbergi P. arabicus P. chabaudi | OW | Middle East, North Africa, Asia | CL |
| Leishmania panamensis | L. panamensis L. gomezi | NW | Panama, Colombia | CL |
| Leishmania equatoriensis | L. hartmanni | NW | Ecuador | CL/MCL |
| Leishmania peruviana | L. verrucarum L. peruensis | NW | Peru | CL |
| Leishmania pifanoi | L. flaviscutellata | NW | Venezuela | CL/DCL |
| Leishmania colombiensis | L. gomezi L hartmanni L. panamensis | NW | Santander, Columbia | CL |
| Leishmania guyanensis | L. anduzei L. umbratilis L. shawi | NW | Brazil | CL |
| Leishmania naiffi | L. ayrozai L. squamiventris | NW | Brazil | CL |
| Leishmania lainsoni | L. ubiquitalis L angelsi | NW | Amanzonas, Ecuador, Peru, Bolivia | CL |
| Leishmania enriettii | L. gomezi L. correalimai | NW | Ghana, Florida, Central Europe | CL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aruleba, R.T.; Carter, K.C.; Brombacher, F.; Hurdayal, R. Can We Harness Immune Responses to Improve Drug Treatment in Leishmaniasis? Microorganisms 2020, 8, 1069. https://doi.org/10.3390/microorganisms8071069
Aruleba RT, Carter KC, Brombacher F, Hurdayal R. Can We Harness Immune Responses to Improve Drug Treatment in Leishmaniasis? Microorganisms. 2020; 8(7):1069. https://doi.org/10.3390/microorganisms8071069
Chicago/Turabian StyleAruleba, Raphael Taiwo, Katharine C. Carter, Frank Brombacher, and Ramona Hurdayal. 2020. "Can We Harness Immune Responses to Improve Drug Treatment in Leishmaniasis?" Microorganisms 8, no. 7: 1069. https://doi.org/10.3390/microorganisms8071069