In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing
2.2. Annotation of the Genome Assemblies
2.3. Selection of LAB
2.4. Selection of Human Microbiomes
2.5. Calculation of Core- and Pan-Genomes
2.6. Calculation of Superpathway Coverage
3. Results and Discussion
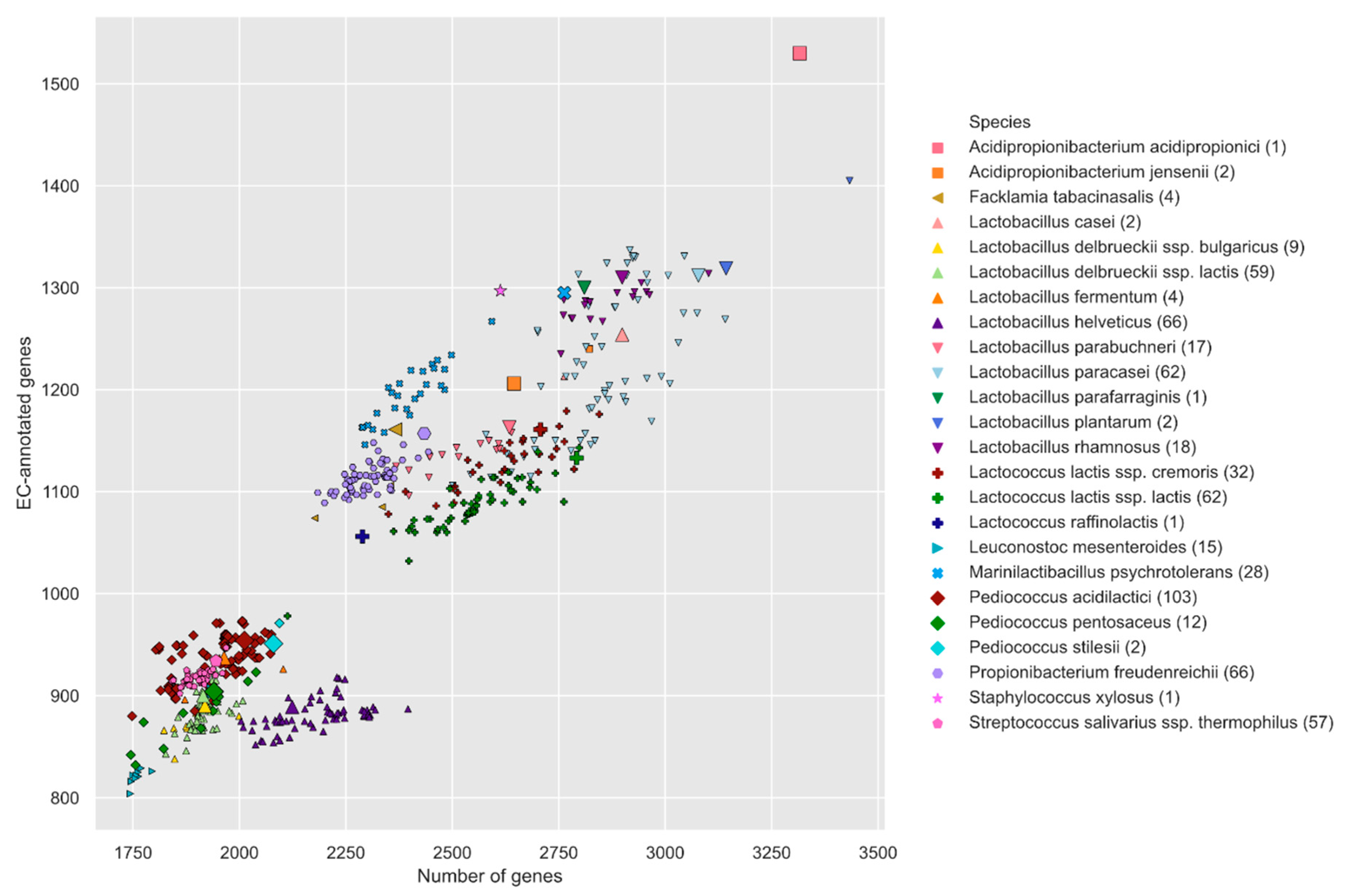
3.1. Liebefeld Collection Overview
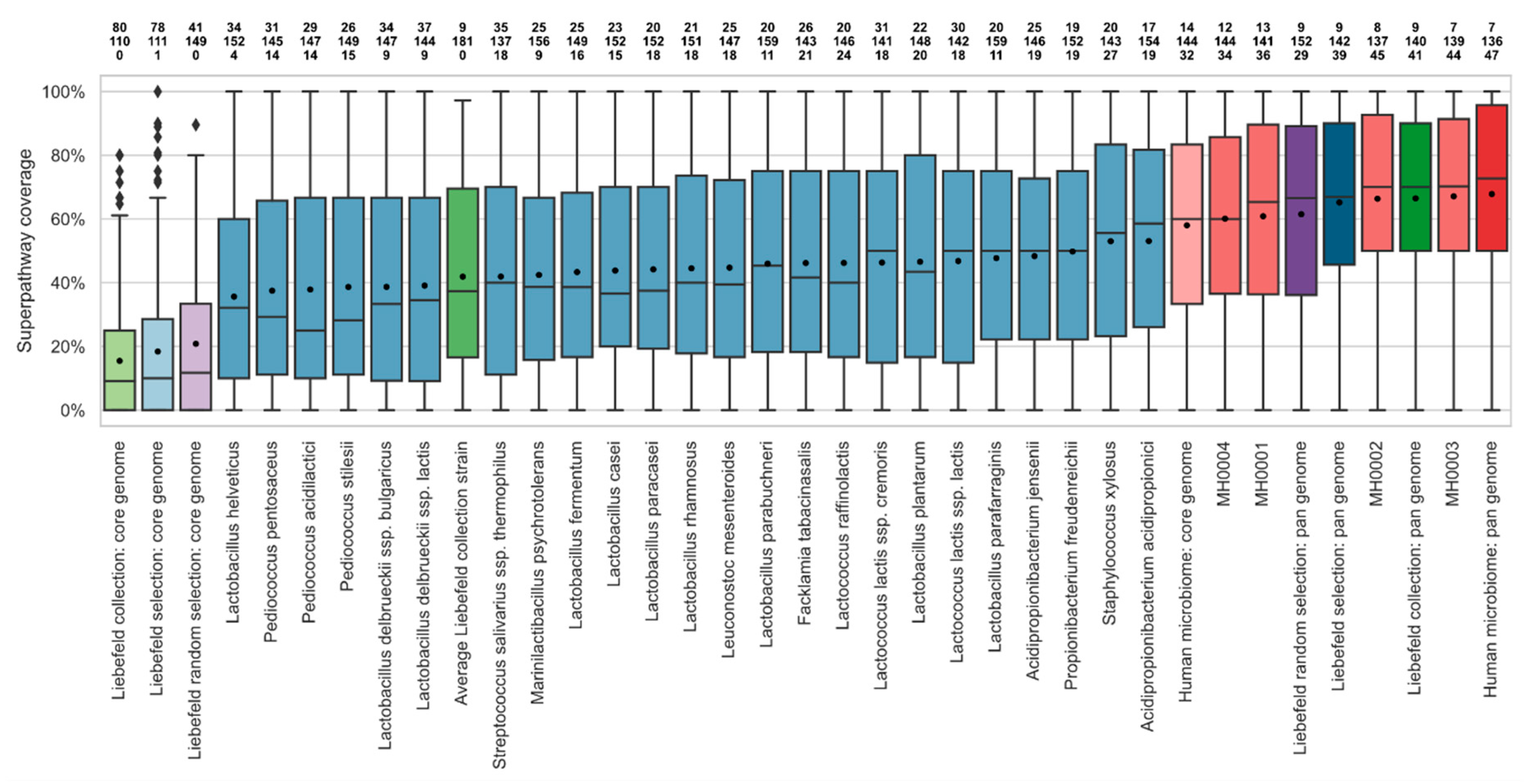
3.2. Comparison of Superpathway Coverage
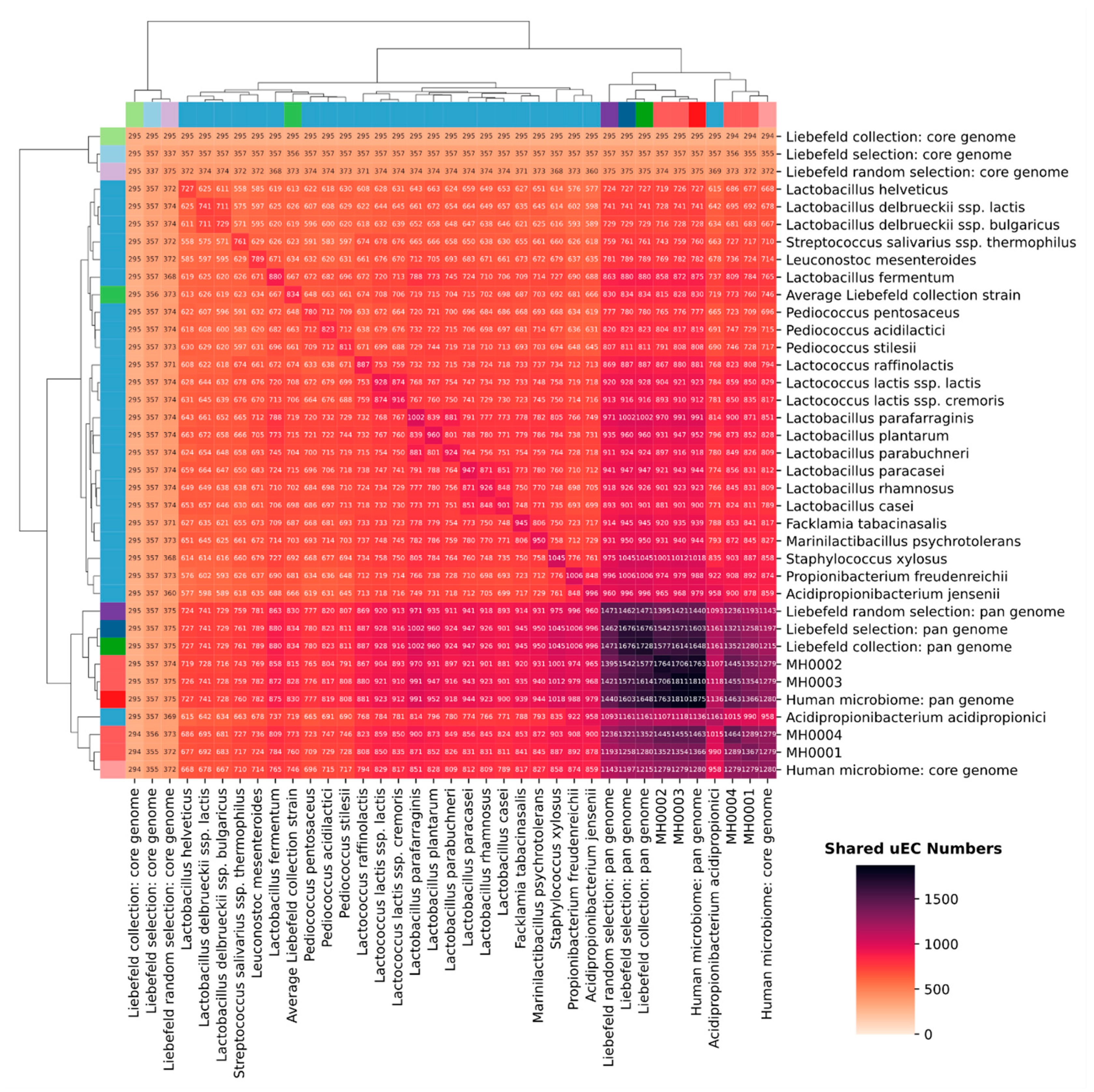
3.3. Comparison of Unique EC Numbers (uECs)
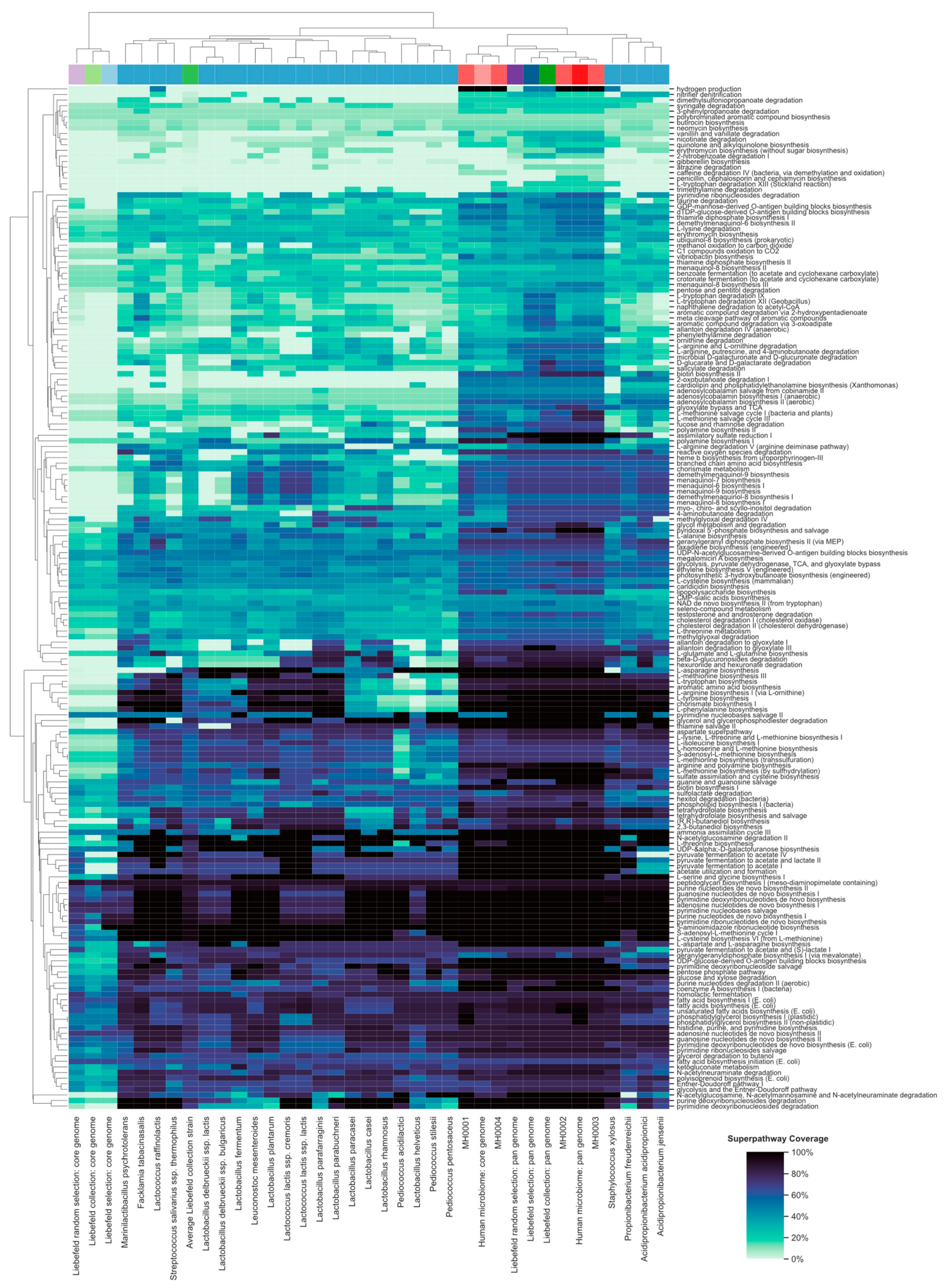
3.4. Functional Properties of the Gut Microbiome, Which Might be Enriched by the Liebefeld Collection
3.4.1. Methylglyoxal Degradation IV Superpathway
3.4.2. Assimilatory Sulfate Reduction I Superpathway
3.4.3. 4-Aminobutanoate Degradation (GABA) Degradation Superpathway
3.4.4. Salicylate Degradation Superpathway
3.5. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morgan, X.C.; Segata, N.; Huttenhower, C. Biodiversity and functional genomics in the human microbiome. Trends Genet. 2013, 29, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, M.; Verma, M.K.; Chauhan, N.S. A review of metabolic potential of human gut microbiome in human nutrition. Arch. Microbiol. 2018, 200, 203–217. [Google Scholar] [CrossRef]
- Balvanera, P.; Pfisterer, A.B.; Buchmann, N.; He, J.S.; Nakashizuka, T.; Raffaelli, D.; Schmid, B. Quantifying the evidence for biodiversity effects on ecosystem functioning and services. Ecol. Lett. 2006, 9, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Dubos, R.; Schaedler, R.W. The digestive tract as an ecological system. Rev. Immunol. Ther. Antimicrob. 1966, 30, 247–252. [Google Scholar] [PubMed]
- Bach, J.-F. The Effect of Infections on Susceptibility to Autoimmune and Allergic Diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef]
- Heiman, M.L.; Greenway, F.L. A healthy gastrointestinal microbiome is dependent on dietary diversity. Mol. Metab. 2016, 5, 317–320. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Metchnikoff, E. Quelques remarques sur le lait aigri. In Revue Générale de Chimie Pure et Appliquée; Bureau de la Revue: Paris, France, 1907; Volume 10, pp. 77–85. [Google Scholar]
- Miquel, S.; Beaumont, M.; Martin, R.; Langella, P.; Braesco, V.; Thomas, M. A proposed framework for an appropriate evaluation scheme for microorganisms as novel foods with a health claim in Europe. Microb. Cell Fact. 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katan, M.B. Why the European Food Safety Authority was right to reject health claims for probiotics. Benef. Microbes 2012, 3, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Salminen, S.; Verhagen, H.; Rowland, I.; Heimbach, J.; Banares, S.; Young, T.; Nomoto, K.; Lalonde, M. Novel probiotics and prebiotics: Road to the market. Curr. Opin. Biotechnol. 2015, 32, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Borresen, E.C.; Henderson, A.J.; Kumar, A.; Weir, T.L.; Ryan, E.P. Fermented foods: Patented approaches and formulations for nutritional supplementation and health promotion. Recent Pat. Food Nutr. Agric. 2012, 4, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; De Filippis, F.; Mauriello, I.E.; Cumbo, F.; Walsh, A.M.; Leech, J.; Cotter, P.D.; Segata, N.; Ercolini, D. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat. Commun. 2020, 11, 2610. [Google Scholar] [CrossRef]
- Li, C.; Bei, T.; Niu, Z.; Guo, X.; Wang, M.; Lu, H.; Gu, X.; Tian, H. Adhesion and Colonization of the Probiotic Lactobacillus rhamnosus Labeled by Dsred2 in Mouse Gut. Curr. Microbiol. 2019, 76, 896–903. [Google Scholar] [CrossRef]
- Xing, Z.; Tang, W.; Yang, Y.; Geng, W.; Rehman, R.U.; Wang, Y. Colonization and Gut Flora Modulation of Lactobacillus kefiranofaciens ZW3 in the Intestinal Tract of Mice. Probiotics Antimicrob. Proteins 2017, 10, 374–382. [Google Scholar] [CrossRef]
- Nemcova, R.; Bomba, A.; Herich, R.; Gancarcikova, S. Colonization capability of orally administered Lactobacillus strains in the gut of gnotobiotic piglets. DTW. Dtsch. Tierarztl. Wochenschr. 1998, 105, 199–200. [Google Scholar]
- Pessione, E. Lactic acid bacteria contribution to gut microbiota complexity: Lights and shadows. Front. Cell Infect. Microbiol. 2012, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.W.; Simpson, J.B.; Roach, J.; Bruno-Barcena, J.M.; Azcarate-Peril, M.A. Prebiotics for Lactose Intolerance: Variability in Galacto-Oligosaccharide Utilization by Intestinal Lactobacillus rhamnosus. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saubade, F.; Hemery, Y.M.; Guyot, J.P.; Humblot, C. Lactic acid fermentation as a tool for increasing the folate content of foods. Crit. Rev. Food Sci. Nutr. 2017, 57, 3894–3910. [Google Scholar] [CrossRef] [PubMed]
- Heeney, D.D.; Gareau, M.G.; Marco, M.L. Intestinal Lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotechnol. 2018, 49, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Dreher, K.; Fulcher, C.A.; Subhraveti, P.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012, 40, D742–D753. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.S.; Altman, T.; Konwar, K.M.; Hanson, N.W.; Kim, D.; Relman, D.A.; Dill, D.L.; Hallam, S.J. GutCyc: A Multi-Study Collection of Human Gut Microbiome Metabolic Models. BioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Shahid, S.U.; Irfan, U. The gut microbiota and its potential role in obesity. Future Microbiol. 2018, 13, 589–603. [Google Scholar]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Wuthrich, D.; Berthoud, H.; Wechsler, D.; Eugster, E.; Irmler, S.; Bruggmann, R. The Histidine Decarboxylase Gene Cluster of Lactobacillus parabuchneri Was Gained by Horizontal Gene Transfer and Is Mobile within the Species. Front. Microbiol. 2017, 8, 218. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Pfrunder, S.; Grossmann, J.; Hunziker, P.; Brunisholz, R.; Gekenidis, M.T.; Drissner, D. Bacillus cereus Group-Type Strain-Specific Diagnostic Peptides. J. Proteome Res. 2016, 15, 3098–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel Wüthrich, R.B. Bacterial Genome Contamination Analysis. Available online: https://github.com/danielwuethrich87/contamination_analysis_of_assembly (accessed on 27 January 2020).
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting biomolecular knowledge via a protein database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the KDD (Knowledge Discovery and Data Mining), Portland, OR, USA, 2–4 August 1996; pp. 226–231. [Google Scholar]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Daniel Wüthrich, R.B. PfastGO. Available online: https://github.com/danielwuethrich87/pfastGO (accessed on 27 January 2020).
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, T.; Ferretti, P.; Maistrenko, O.M.; Bork, P. Diversity within species: Interpreting strains in microbiomes. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; et al. 1520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019, 37, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Altman, T.; Travers, M.; Kothari, A.; Caspi, R.; Karp, P.D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinform. 2013, 14, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roder, T. MetaCycSuperPathwayParser. Available online: https://binfgitlab.unibe.ch/troder/metacycsuperpathwayparser (accessed on 3 March 2020).
- Li, Q.; Chen, H.; Zhang, M.; Wu, T.; Liu, R.; Zhang, Z. Potential Correlation between Dietary Fiber-Suppressed Microbial Conversion of Choline to Trimethylamine and Formation of Methylglyoxal. J. Agric. Food Chem. 2020, 67, 13247–13257. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Q.; Jiang, H. Gut Microbiota in Atherosclerosis: Focus on Trimethylamine N-Oxide. APMIS 2020. [Google Scholar] [CrossRef] [Green Version]
- Burton, K.J.; Kruger, R.; Scherz, V.; Munger, L.H.; Picone, G.; Vionnet, N.; Bertelli, C.; Greub, G.; Capozzi, F.; Vergeres, G. Trimethylamine-N-Oxide Postprandial Response in Plasma and Urine Is Lower After Fermented Compared to Non-Fermented Dairy Consumption in Healthy Adults. Nutrients 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Kushkevych, I.; Cejnar, J.; Treml, J.; Dordevic, D.; Kollar, P.; Vitezova, M. Recent Advances in Metabolic Pathways of Sulfate Reduction in Intestinal Bacteria. Cells 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Kushkevych, I.; Kotrsova, V.; Dordevic, D.; Bunkova, L.; Vitezova, M.; Amedei, A. Hydrogen Sulfide Effects on the Survival of Lactobacilli with Emphasis on the Development of Inflammatory Bowel Diseases. Biomolecules 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, G.V.; Choi, K.; Klemashevich, C.; Wu, C.; Prabakaran, D.; Pan, L.B.; Steinmeyer, S.; Mueller, C.; Yousofshahi, M.; Alaniz, R.C.; et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat. Commun. 2014, 5, 5492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, K.W. Aryl hydrocarbon receptor (AHR): From selected human target genes and crosstalk with transcription factors to multiple AHR functions. Biochem. Pharmacol. 2019, 168, 65–70. [Google Scholar] [CrossRef]
- Damman, C.J. Salicylates and the Microbiota: A New Mechanistic Understanding of an Ancient Drug’s Role in Dermatological and Gastrointestinal Disease. Drug Dev. Res. 2013, 74, 344–352. [Google Scholar] [CrossRef]
- Alp, D.; Kuleasan, H. Adhesion mechanisms of lactic acid bacteria: Conventional and novel approaches for testing. World J. Microbiol. Biotechnol. 2019, 35, 156. [Google Scholar] [CrossRef]
- Nishiyama, K.; Sugiyama, M.; Mukai, T. Adhesion Properties of Lactic Acid Bacteria on Intestinal Mucin. Microorganisms 2016, 4. [Google Scholar] [CrossRef]
- Rajilic-Stojanovic, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- De Paepe, K.; Verspreet, J.; Rezaei, M.N.; Hidalgo Martinez, S.; Meysman, F.; Van de Walle, D.; Dewettinck, K.; Raes, J.; Courtin, C.; Van de Wiele, T. Isolation of wheat bran-colonizing and metabolizing species from the human fecal microbiota. PeerJ 2019, 7, e6293. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, M.; Zhao, X.; Li, X. Effects of garlic polysaccharide on alcoholic liver fibrosis and intestinal microflora in mice. Pharm. Biol. 2018, 56, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Rocha, E.P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Mahowald, M.A.; Ley, R.E.; Lozupone, C.A.; Hamady, M.; Martens, E.C.; Henrissat, B.; Coutinho, P.M.; Minx, P.; Latreille, P.; et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007, 5, e156. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roder, T.; Wüthrich, D.; Bär, C.; Sattari, Z.; von Ah, U.; Ronchi, F.; Macpherson, A.J.; Ganal-Vonarburg, S.C.; Bruggmann, R.; Vergères, G. In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes. Microorganisms 2020, 8, 966. https://doi.org/10.3390/microorganisms8070966
Roder T, Wüthrich D, Bär C, Sattari Z, von Ah U, Ronchi F, Macpherson AJ, Ganal-Vonarburg SC, Bruggmann R, Vergères G. In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes. Microorganisms. 2020; 8(7):966. https://doi.org/10.3390/microorganisms8070966
Chicago/Turabian StyleRoder, Thomas, Daniel Wüthrich, Cornelia Bär, Zahra Sattari, Ueli von Ah, Francesca Ronchi, Andrew J. Macpherson, Stephanie C. Ganal-Vonarburg, Rémy Bruggmann, and Guy Vergères. 2020. "In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes" Microorganisms 8, no. 7: 966. https://doi.org/10.3390/microorganisms8070966
APA StyleRoder, T., Wüthrich, D., Bär, C., Sattari, Z., von Ah, U., Ronchi, F., Macpherson, A. J., Ganal-Vonarburg, S. C., Bruggmann, R., & Vergères, G. (2020). In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes. Microorganisms, 8(7), 966. https://doi.org/10.3390/microorganisms8070966