Effect of Sample Collection (Manual Expression vs. Pumping) and Skimming on the Microbial Profile of Human Milk Using Culture Techniques and Metataxonomic Analysis

 and
and
Abstract
:1. Introduction
2. Materials and Methods
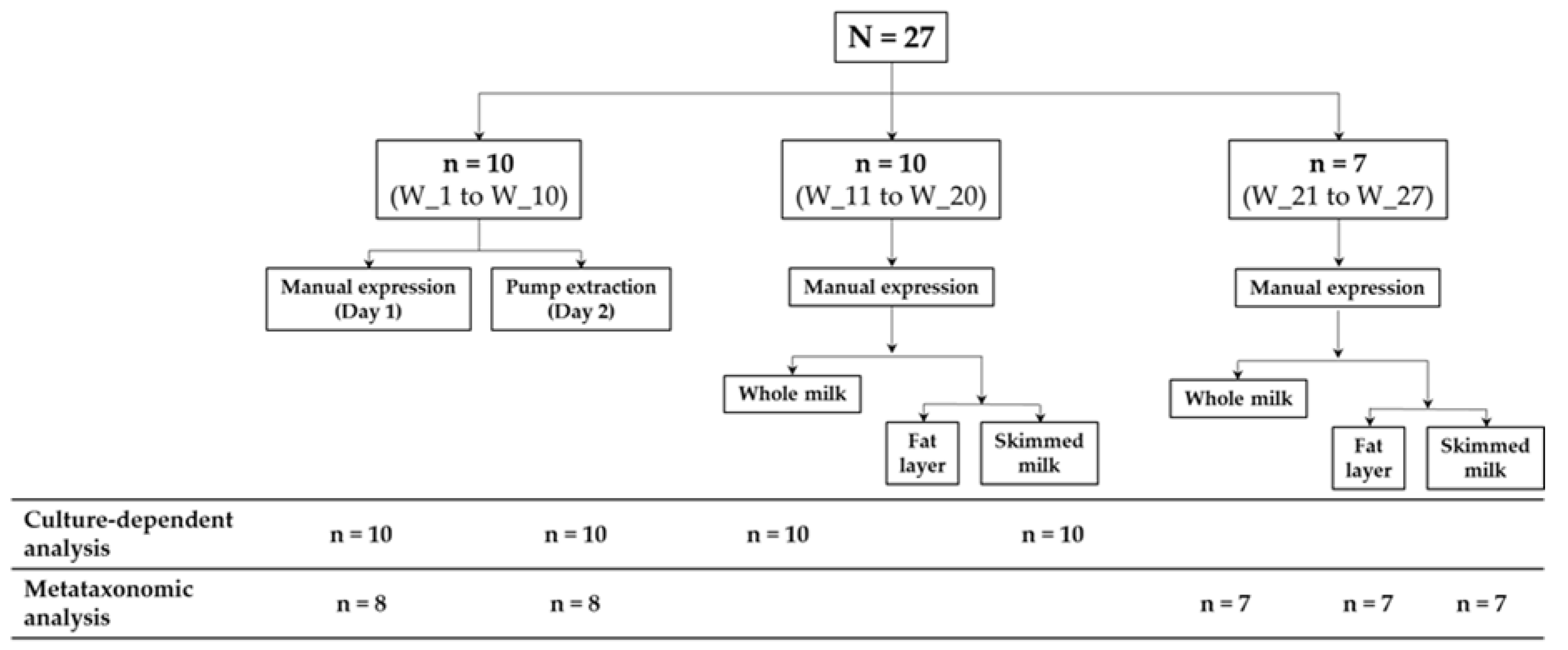
2.1. Participating Women and Collection of the Samples
2.2. Cultures and Identification of Isolates
2.3. DNA Extraction from the Milk Samples
2.4. PCR Amplification and Sequencing
2.5. Bioinformatic and Statistical Analysis
3. Results
3.1. Culture-Dependent Analysis of the Samples: Effect of the Collection Method
3.2. Culture-Dependent Analysis of the Samples: Effect of Skimming
3.3. Microbiome Analysis of the Samples: Effect of the Collection Method
3.4. Microbiome Analysis of the Samples: Effect of Skimming
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fernández, L.; Langa, S.; Martín, V.; Maldonado, A.; Jiménez, E.; Martín, R.; Rodríguez, J.M. The human milk microbiota: Origin and potential roles in health and disease. Pharmacol. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef]
- Fernández, L.; Arroyo, R.; Espinosa, I.; Marín, M.; Jiménez, E.; Rodríguez, J.M. Probiotics for human lactational mastitis. Benef. Microbes 2014, 5, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gallego, C.; Garcia-Mantrana, I.; Salminen, S.; Collado, M.C. The human milk microbiome and factors influencing its composition and activity. Semin. Fetal Neonatal Med. 2016, 21, 400–405. [Google Scholar] [CrossRef] [Green Version]
- LaTuga, M.S.; Stuebe, A.; Seed, P.C. A review of the source and function of microbiota in breast milk. Semin. Reprod. Med. 2014, 32, 68–73. [Google Scholar] [PubMed] [Green Version]
- Arroyo, R.; Mediano, P.; Martín, V.; Jiménez, E.; Delgado, S.; Fernández, L.; Marín, M.; Rodríguez, J.M. Diagnóstico etiológico de las mastitis infecciosas: Propuesta de protocolo para el cultivo de muestras de leche humana [Etiological diagnosis of infectious mastitis: Proposal of a protocol for the culture of human milk samples]. Acta Pediatr. Esp. 2011, 69, 276–281. [Google Scholar]
- Delgado, S.; García-Garrote, F.; Padilla, B.; Rodríguez, J.M.; Romero, B. Microbiological diagnosis of bacterial infection associated with delivery and postpartum. In Procedures in Clinical Microbiology; Number 54; Cercenado, E., Cantón, R., Eds.; Spanish Society of Infectious Diseases and Clinical Microbiology: Madrid, Spain, 2015. [Google Scholar]
- Brown, S.L.; Bright, R.A.; Dwyer, D.E.; Foxman, B. Breast pump adverse events: Reports to the Food and Drug Administration. J. Hum. Lact. 2005, 21, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.L.; Arroyo, R.; Jiménez, E.; Gómez, A.; Fernández, L.; Rodríguez, J.M. Cold storage of human milk: Effect on its bacterial composition. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Flores, B.-A.; Martín, J.-C.; Morante, M.A.-S.; García, N.R.-L.; Sierra, G.-C.; De la Cruz, J.-B.; Martín, C.-A.-A.; Escuder-Vieco, D.; Soriano, M.-R.; Chaves, F.; et al. Comparison of two methods for cleaning breast pump milk collection kits in human milk banks. J. Hosp. Infect. 2019, 103, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, E.; Arroyo, R.; Cárdenas, N.; Marín, M.; Serrano, P.; Fernández, L.; Rodríguez, J.M. Mammary candidiasis: A medical condition without scientific evidence? PLoS ONE 2017, 12, e0181071. [Google Scholar] [CrossRef]
- Paviour, S.; Musaad, S.; Roberts, S.; Taylor, G.; Taylor, S.; Shore, K.; Lang, S.; Holland, D. Corynebacterium species isolated from patients with mastitis. Clin. Infect. Dis. 2002, 35, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Marín, M.; Arroyo, R.; Espinosa-Martos, I.; Fernández, L.; Rodríguez, J.M. Identification of Emerging human mastitis pathogens by MALDI-TOF and assessment of their antibiotic resistance patterns. Front. Microbiol. 2017, 8, 1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Hunt, K.M.; Foster, J.A.; Forney, L.J.; Schütte, U.M.; Beck, D.L.; Abdo, Z.; Fox, L.K.; Williams, J.E.; McGuire, M.K.; McGuire, M.A. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 2011, 6, e21313. [Google Scholar] [CrossRef] [Green Version]
- Lackey, K.A.; Williams, J.E.; Meehan, C.L.; Zachek, J.A.; Benda, E.D.; Price, W.J.; Foster, J.A.; Sellen, D.W.; Kamau-Mbuthia, E.W.; Kamundia, E.W.; et al. What’s normal? Microbiomes in human milk and infant feces are related to each other but vary geographically: The INSPIRE study. Front. Nutr. 2019, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, E.; Delgado, S.; Maldonado, A.; Arroyo, R.; Albujar, M.; García, N.; Jariod, M.; Fernández, L.; Gómez, A.; Rodríguez, J.M. Staphylococcus epidermidis: A differential trait of the fecal microbiota of breast-fed infants. BMC Microbiol. 2008, 8, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solís, G.; de los Reyes-Gavilan, C.G.; Fernández, N.; Margolles, A.; Gueimonde, M. Establishment and development of lactic acid bacteria and bifidobacteria microbiota in breast-milk and the infant gut. Anaerobe 2010, 16, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Schanler, R.J.; Fraley, J.K.; Lau, C.; Hurst, N.M.; Hovarth, L.; Rossmann, S.N. Breastmilk cultures and infection in extremely premature infants. J. Perinatol. 2011, 31, 335–338. [Google Scholar] [CrossRef] [Green Version]
- Martín, R.; Langa, S.; Reviriego, C.; Jiménez, E.; Marín, M.L.; Xaus, J.; Fernández, L.; Rodríguez, J.M. Human milk is a source of lactic acid bacteria for the infant gut. J. Pediatr. 2003, 143, 754–758. [Google Scholar] [CrossRef]
- Abrahamsson, T.R.; Sinkiewicz, G.; Jakobsson, T.; Fredrikson, M.; Björkstén, B. Probiotic lactobacilli in breast milk and infant stool in relation to oral intake during the first year of life. J. Ped. Gastroenterol. Nutr. 2009, 49, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, R.; Jiménez, E.; Heilig, H.; Fernández, L.; Marín, M.L.; Zoetendal, E.G.; Rodriguez, J.M. Isolation of bifidobacteria from breast milk and assessment of the bifidobacterial population by PCR-denaturing gradient gel electrophoresis and quantitative real-time PCR. Appl. Environ. Microbiol. 2009, 75, 965–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboleya, S.; Ruas-Madiedo, P.; Margolles, A.; Solís, G.; Salminen, S.; de los Reyes-Gavilán, C.G.; Gueimonde, M. Characterization and in vitro properties of potentially probiotic Bifidobacterium strains isolated from breast-milk. Int. J. Food Microbiol. 2011, 149, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.; Curley, D.; O’Callaghan, T.F.; O’Shea, C.-A.; Dempsey, E.M.; O’Toole, P.W.; Ross, R.P.; Ryan, C.A.; Stanton, C. The composition of human milk and infant fecal microbiota over the first three months of life: A pilot study. Sci. Rep. 2017, 7, 40597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerin, J.; Burgain, J.; Gomand, F.; Scher, J.; Gaiani, C. Milk fat globule membrane glycoproteins: Valuable ingredients for lactic acid bacteria encapsulation? Crit. Rev. Food Sci. Nutr. 2019, 59, 639–651. [Google Scholar] [CrossRef]
- Douëllou, T.; Montel, M.C.; Thevenot Sergentet, D. Anti-adhesive properties of bovine oligosaccharides and bovine milk fat globule membrane-associated glycoconjugates against bacterial food enteropathogens. J. Dairy Sci. 2017, 100, 3348–3359. [Google Scholar] [CrossRef]
- McGuire, M.K.; McGuire, M.A. Human milk: Mother nature’s prototypical probiotic food? Adv. Nutr. 2015, 6, 112–123. [Google Scholar] [CrossRef]
- McGuire, M.K.; McGuire, M.A. Got bacteria? The astounding, yet not-so-surprising, microbiome of human milk. Curr. Opin. Biotechnol. 2017, 44, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Rapin, A.; Pattaroni, C.; Marsland, B.J.; Harris, N.L. Microbiota analysis using an Illumina MiSeq platform to sequence 16S rRNA genes. Curr. Protoc. Mouse Biol. 2017, 7, 100–129. [Google Scholar] [CrossRef]
- Tawfik, S.A.; Azab, M.M.; Ahmed, A.A.A.; Fayyad, D.M. Illumina MiSeq sequencing for preliminary analysis of microbiome causing primary endodontic infections in Egypt. Int. J. Microbiol. 2018, 2018, 2837328. [Google Scholar] [CrossRef] [Green Version]
- Dahlberg, J.; Sun, L.; Persson Waller, K.; Östensson, K.; McGuire, M.; Agenäs, S.; Dicksved, J. Microbiota data from low biomass milk samples is markedly affected by laboratory and reagent contamination. PLoS ONE 2019, 14, e0218257. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Woman | Sample Collection 1 | Agar Media 2 | Bacterial Counts 3 | Bacterial Species 4 |
|---|---|---|---|---|
| W_1 | Manual | CNA | 3.72 ± 0.13 | S. epidermidis; Corynebacterium spp. |
| MRS-Cys | 3.38 ± 0.06 | S. epidermidis; Lc. lactis | ||
| MCK | 3.30 ± 0.13 | A. ursingii | ||
| Pump | CNA | 3.51 ± 0.04 | S. epidermidis; Corynebacterium spp. | |
| (SUD) | MRS-Cys | 3.34 ± 0.16 | S. epidermidis; Lc. lactis | |
| MCK | 3.86 ± 0.08 | A. johnsonii | ||
| W_2 | Manual | CNA | 3.00 ± 0.04 | S. epidermidis |
| MRS-Cys | 3.47 ± 0.14 | S. epidermidis | ||
| MCK | n.d. 5 | - | ||
| Pump | CNA | 3.08 ± 0.11 | S. epidermidis | |
| (SUD) | MRS-Cys | 2.98 ± 0.11 | S. epidermidis | |
| MCK | n.d. | - | ||
| W_3 | Manual | CNA | n.d. | - |
| MRS-Cys | n.d. | - | ||
| MCK | n.d. | - | ||
| Pump | CNA | n.d. | - | |
| (SUD) | MRS-Cys | n.d. | - | |
| MCK | n.d. | - | ||
| W_4 | Manual | CNA | n.d. | - |
| MRS-Cys | n.d. | - | ||
| MCK | n.d. | - | ||
| Pump | CNA | 2.34 ± 0.10 | S. epidermidis | |
| (SUD) | MRS-Cys | 2.30 ± 0.11 | S. epidermidis | |
| MCK | n.d. | - | ||
| W_5 | Manual | CNA | 2.60 ± 0.16 | S. epidermidis |
| MRS-Cys | 3.00 ± 0.16 | S. epidermidis | ||
| MCK | n.d. | - | ||
| Pump | CNA | 2.78 ± 0.11 | S. epidermidis | |
| (SUD) | MRS-Cys | 2.60 ± 0.10 | S. epidermidis | |
| MCK | n.d. | - | ||
| W_6 | Manual | CNA | n.d. | - |
| MRS-Cys | 2.64 ± 0.20 | S. epidermidis; Str. parasanguinis/lactarius; L. salivarius | ||
| MCK | n.d. | - | ||
| Pump | CNA | 2.86 ± 0.07 | S. epidermidis; Str. parasanguinis/lactarius | |
| (SUD) | MRS-Cys | 2.58 ± 0.08 | S. epidermidis; L. salivarius | |
| MCK | n.d. | - | ||
| W_7 | Manual | CNA | 3.48 ± 0.16 | S. epidermidis; Ko. kristinae |
| MRS-Cys | 3.31 ± 0.13 | S. epidermidis; Li. reuteri | ||
| MCK | n.d. | - | ||
| Pump | CNA | 3.33 ± 0.17 | S. epidermidis; Ko. kristinae | |
| (SUD) | MRS-Cys | 3.16 ± 0.21 | S. epidermidis; Li. reuteri | |
| MCK | n.d. | - | ||
| W_8 | Manual | CNA | 3.12 ± 0.21 | S. epidermidis; Str. salivarius |
| MRS-Cys | 2.30 ± 0.13 | S. epidermidis | ||
| MCK | 2.79 ± 0.05 | A. ursingii | ||
| Pump | CNA | 4.03 ± 0.11 | S. epidermidis; Str. salivarius | |
| (OMP) | MRS-Cys | 3.88 ± 0.12 | S. epidermidis | |
| MCK | 2.26 ± 0.07 | A. ursingii | ||
| W_9 | Manual | CNA | 2.76 ± 0.07 | S. epidermidis; Str. parasanguinis/lactarius; Ko. rhizophila |
| MRS-Cys | 2.60 ± 0.16 | S. epidermidis | ||
| MCK | n.d. | - | ||
| Pump | CNA | 3.26 ± 0.14 | S. epidermidis; Str. parasanguinis/lactarius; Ko. rhizophila | |
| (SUD) | MRS-Cys | 2.30 ± 0.08 | S. epidermidis | |
| MCK | n.d. | - | ||
| W_10 | Manual | CNA | n.d. | - |
| MRS-Cys | n.d. | - | ||
| MCK | n.d. | - | ||
| Pump | CNA | 4.38 ± 0.18 | Stn. maltophilia | |
| (OMP) | MRS-Cys | n.d. | - | |
| MCK | 4.87 ± 0.09 | K. oxytoca; Stn. maltophilia |
| Woman | Agar Media 1 | Fraction | Bacterial Counts 2 | Bacterial Species 3 |
|---|---|---|---|---|
| W_11 | CNA | Whole milk | 2.48 ± 0.16 | S. epidermidis |
| Skimmed milk | 2.46 ± 0.11 | S. epidermidis | ||
| MRS-Cys | Whole milk | 2.30 ± 0.18 | S. epidermidis | |
| Skimmed milk | 2.34 ± 0.16 | S. epidermidis | ||
| W_12 | CNA | Whole milk | 2.57 ± 0.17 | S. epidermidis; Str. salivarius |
| Skimmed milk | 2.66 ± 0.10 | S. epidermidis; Str. salivarius | ||
| W_12 | MRS-Cys | Whole milk | 2.60 ± 0.08 | S. epidermidis |
| Skimmed milk | 2.70 ± 0.11 | S. epidermidis | ||
| W_13 | CNA | Whole milk | n.d. 4 | - |
| Skimmed milk | n.d. | - | ||
| MRS-Cys | Whole milk | n.d. | - | |
| Skimmed milk | n.d. | - | ||
| W_14 | CNA | Whole milk | 2.58 ± 0.13 | S. epidermidis |
| Skimmed milk | 2.54 ± 0.17 | S. epidermidis | ||
| MRS-Cys | Whole milk | 2.42 ± 0.14 | S. epidermidis | |
| Skimmed milk | 2.38 ± 0.16 | S. epidermidis | ||
| W_15 | CNA | Whole milk | 2.38 ± 0.13 | Str. mitis; K. kristinae |
| Skimmed milk | 2.67 ± 0.14 | Str. mitis; K. kristinae | ||
| MRS-Cys | Whole milk | 2.56 ± 0.11 | S. epidermidis; L. salivarius | |
| Skimmed milk | 2.69 ± 0.13 | S. epidermidis; L. salivarius | ||
| W_16 | CNA | Whole milk | 2.70 ± 0.14 | S. epidermidis; Corynebacterium spp. |
| Skimmed milk | 2.73 ± 0.18 | S. epidermidis; Corynebacterium spp. | ||
| MRS-Cys | Whole milk | 2.51 ± 0.16 | S. epidermidis | |
| Skimmed milk | 2.40 ± 0.21 | S. epidermidis | ||
| W_17 | CNA | Whole milk | 2.15 ± 0.11 | S. epidermidis; E. faecium |
| Skimmed milk | 2.04 ± 0.06 | S. epidermidis; E. faecium | ||
| MRS-Cys | Whole milk | n.d. | - | |
| Skimmed milk | n.d. | - | ||
| W_18 | CNA | Whole milk | 2.72 ± 0.06 | S. epidermidis; R. mucilaginosa |
| Skimmed milk | 2.68 ± 0.14 | S. epidermidis; R. mucilaginosa | ||
| MRS-Cys | Whole milk | 2.48 ± 0.11 | S. epidermidis | |
| Skimmed milk | 2.44 ± 0.13 | S. epidermidis | ||
| W_19 | CNA | Whole milk | 2.40 ± 0.17 | S. epidermidis |
| Skimmed milk | 2.36 ± 0.10 | S. epidermidis | ||
| MRS-Cys | Whole milk | 2.34 ± 0.16 | S. epidermidis; Lc. lactis | |
| Skimmed milk | 2.41 ± 0.17 | S. epidermidis; Lc. lactis | ||
| W_20 | CNA | Whole milk | n.d. | - |
| Skimmed milk | n.d. | - | ||
| MRS-Cys | Whole milk | n.d. | - | |
| Skimmed milk | n.d. | - |
| Phylum/Genus | Manual Extraction | Pumping | |||
|---|---|---|---|---|---|
| n (%) | Relative Abundance | n (%) | Relative Abundance | p-Value 1 | |
| Firmicutes | |||||
| Streptococcus | 8 (100%) | 23.94 (14.22–42.83) | 8 (100%) | 37.49 (10.13–55.44) | 0.88 |
| Staphylococcus | 8 (100%) | 20.36 (2.74–22.82) | 8 (100%) | 8.26 (2.37–14.22) | 0.16 |
| Veillonella | 5 (63%) | 0.21 (0.00–1.44) | 7 (88%) | 0.86 (0.25–2.51) | 0.40 |
| Gemella | 4 (50%) | 0.10 (0.00–0.40) | 6 (75%) | 0.48 (0.20–0.68) | 0.36 |
| Paenibacillus | 8 (100%) | 0.19 (0.17–0.20) | 8 (100%) | 0.14 (0.11–0.16) | 0.17 |
| Clostridium | 8 (100%) | 0.13 (0.09–0.20) | 6 (75%) | 0.17 (0.11–0.19) | 1.00 |
| Bacillus | 7 (88%) | 0.18 (0.05–0.39) | 7 (88%) | 0.07 (0.06–0.14) | 0.56 |
| Granulicatella | 1 (13%) | <0.00 | 4 (50%) | 0.03 (0.00–0.27) | 0.10 |
| Leuconostoc | 1 (13%) | <0.00 | 4 (50%) | 0.03 (0.00–0.17) | 0.11 |
| Actinobacteria | |||||
| Rothia | 8 (100%) | 3.27 (0.72–6.51) | 8 (100%) | 1.60 (1.26-4.30) | 0.75 |
| Corynebacterium | 8 (100%) | 0.39 (0.28-–0.73) | 7 (88%) | 0.16 (0.08-1.04) | 0.44 |
| Cellulomonas | 4 (50%) | 0.13 (0.00–0.37) | 5 (63%) | 0.09 (0.00-0.22) | 0.87 |
| Streptomyces | 4 (50%) | 0.09 (0.00–0.26) | 5 (63%) | 0.13 (0.00-0.17) | 0.87 |
| Actinomyces | 5 (63%) | 0.06 (0.00–0.20) | 4 (50%) | 0.06 (0.00-0.54) | 0.87 |
| Proteobacteria | |||||
| Acinetobacter | 5 (63%) | 0.34 (0.00–2.07) | 6 (75%) | 0.28 (0.05–1.00) | 0.87 |
| Sphingomonas | 6 (75%) | 0.34 (0.05–0.86) | 4 (50%) | 0.02 (0.00–0.12) | 0.21 |
| Pseudomonas | 6 (75%) | 0.09 (0.03–0.42) | 7 (88%) | 0.58 (0.15–1.18) | 0.23 |
| Erwinia | 2 (25%) | 0.00 (0.00–0.05) | 3 (38%) | 0.00 (0.00–0.13) | 0.65 |
| Unclassified genera | 8 (100%) | 19.49 (14.23–27.96) | 8 (100%) | 15.82 (14.18–28.15) | 0.80 |
| Phylum/Genus | Whole Milk | Fat Layer | Skimmed Milk | ||||
|---|---|---|---|---|---|---|---|
| n (%) | Relative Abundance 1 | n (%) | Relative Abundance | n (%) | Relative Abundance | p-Value 2 | |
| Firmicutes | |||||||
| Staphylococcus | 7 (100%) | 12.10 (7.95–25.17) | 7 (100%) | 3.43 (1.23–16.61) | 7 (100%) | 9.61 (0.94–21.08) | 0.53 |
| Streptococcus | 7 (100%) | 2.31 (1.29–4.57) | 7 (100%) | 4.03 (2.54–18.01) | 7 (100%) | 2.66 (2.33–4.95) | 0.59 |
| Lactobacillus | 7 (100%) | 1.43 (0.78–2.14) | 7 (100%) | 1.87 (0.86–3.78) | 7 (100%) | 2.20 (0.46–3.55) | 0.95 |
| Pediococcus | 7 (100%) | 1.31 (0.87–1.84) | 6 (86%) | 1.38 (0.78–1.49) | 6 (86%) | 1.39 (0.66–1.85) | 0.82 |
| Bacillus | 6 (86%) | 0.34 (0.24–0.45) | 4 (57%) | 0.17 (0.00–0.29) | 5 (71%) | 0.33 (0.16–0.44) | 0.39 |
| Peptoniphilus | 5 (71%) | 0.35 (0.11–1.44) | 5 (71%) | 0.81 (0.40–1.27) | 3 (43%) | 0.00 (0.00–0.84) | 0.56 |
| Lactococcus | 3 (43%) | 0.00 (0.00–2.91) | 4 (57%) | 0.22 (0.00–6.98) | 6 (86%) | 3.57 (1.76–7.09) | 0.33 |
| Actinobacteria | |||||||
| Propionibacterium | 5 (71%) | 1.11 (0.12–4.98) | 6 (86%) | 6.52 (0.60–7.08) | 6 (86%) | 1.37 (1.14–2.35) | 0.84 |
| Corynebacterium | 5 (71%) | 1.11 (0.23–2.52) | 3 (43%) | 0.00 (0.00–0.84) | 4 (57%) | 0.48 (0.00–3.13) | 0.57 |
| Bifidobacterium | 2 (29%) | 0.00 (0.00–0.66) | 3 (43%) | 0.00 (0.00–0.33) | 2 (29%) | 0.00 (0.00–0.14) | 0.84 |
| Proteobacteria | |||||||
| Chondromyces | 5 (71%) | 2.51 (0.99–6.15) | 5 (71%) | 3.43 (1.23–3.80) | 4 (57%) | 1.31 (0.00–4.18) | 0.79 |
| Burkholderia | 7 (100%) | 0.77 (0.64–0.86) | 7 (100%) | 1.40 (1.06–1.60) | 6 (86%) | 1.03 (0.62–1.12) | 0.04 |
| Sphingomonas | 5 (71%) | 0.60 (0.12–3.32) | 6 (86%) | 0.94 (0.41–1.46) | 4 (57%) | 0.38 (0.00–0.75) | 0.38 |
| Hyphomicrobium | 5 (71%) | 0.30 (0.11–0.68) | 6 (86%) | 0.43 (0.30–0.62) | 3 (43%) | 0.00 (0.00–0.37) | 0.28 |
| Novosphingobium | 2 (29%) | 0.00 (0.00–0.21) | 3 (43%) | 0.00 (0.00–0.37) | 4 (57%) | 0.13 (0.00–0.52) | 0.69 |
| Azospirillum | 2 (29%) | 0.00 (0.00–0.18) | 4 (57%) | 0.38 (0.00–0.68) | 4 (57%) | 0.43 (0.00–1.47) | 0.25 |
| Chondromyces | 5 (71%) | 2.51 (0.99–6.15) | 5 (71%) | 3.43 (1.23–3.80) | 4 (57%) | 1.31 (0.00–4.18) | 0.79 |
| Bacteroidetes | |||||||
| Pedobacter | 7 (100%) | 0.61 (0.38–0.88) | 7 (100%) | 0.56 (0.42–0.83) | 6 (86%) | 0.72 (0.44–0.89) | 0.96 |
| Unclassified genera | 7 (100%) | 27.7 (23.86–31.55) | 7 (100%) | 31.81 (28.09–34.53) | 7 (100%) | 30.82 (26.71–35.82) | 0.66 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Cruz, M.; Alba, C.; Aparicio, M.; Checa, M.Á.; Fernández, L.; Rodríguez, J.M. Effect of Sample Collection (Manual Expression vs. Pumping) and Skimming on the Microbial Profile of Human Milk Using Culture Techniques and Metataxonomic Analysis. Microorganisms 2020, 8, 1278. https://doi.org/10.3390/microorganisms8091278
Rodríguez-Cruz M, Alba C, Aparicio M, Checa MÁ, Fernández L, Rodríguez JM. Effect of Sample Collection (Manual Expression vs. Pumping) and Skimming on the Microbial Profile of Human Milk Using Culture Techniques and Metataxonomic Analysis. Microorganisms. 2020; 8(9):1278. https://doi.org/10.3390/microorganisms8091278
Chicago/Turabian StyleRodríguez-Cruz, Maricela, Claudio Alba, Marina Aparicio, María Ángeles Checa, Leonides Fernández, and Juan Miguel Rodríguez. 2020. "Effect of Sample Collection (Manual Expression vs. Pumping) and Skimming on the Microbial Profile of Human Milk Using Culture Techniques and Metataxonomic Analysis" Microorganisms 8, no. 9: 1278. https://doi.org/10.3390/microorganisms8091278