A Novel Primer Mixture for GH48 Genes: Quantification and Identification of Truly Cellulolytic Bacteria in Biogas Fermenters

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Primer Development
2.2. Nucleic Acid Extraction from Biogas Fermenter Samples
2.3. (Quantitative) Polymerase Chain Reaction (qPCR)
2.4. Reverse Transcription (RT)
2.5. DNA Standard for Absolute Quantification
2.6. GH48 Reference Database Construction
2.7. Phylogenetic Correlation of 16S rRNA Genes to GH48 Gene Sequences
2.8. Next-Generation-Sequencing (NGS) and Data Analysis
3. Results
3.1. Primer Development
3.2. Isolation of Metagenomic DNA and Metatranscriptomic RNA
3.3. Quantification of GH48 and 16S rRNA Gene Sequences
3.4. Reference Database Construction
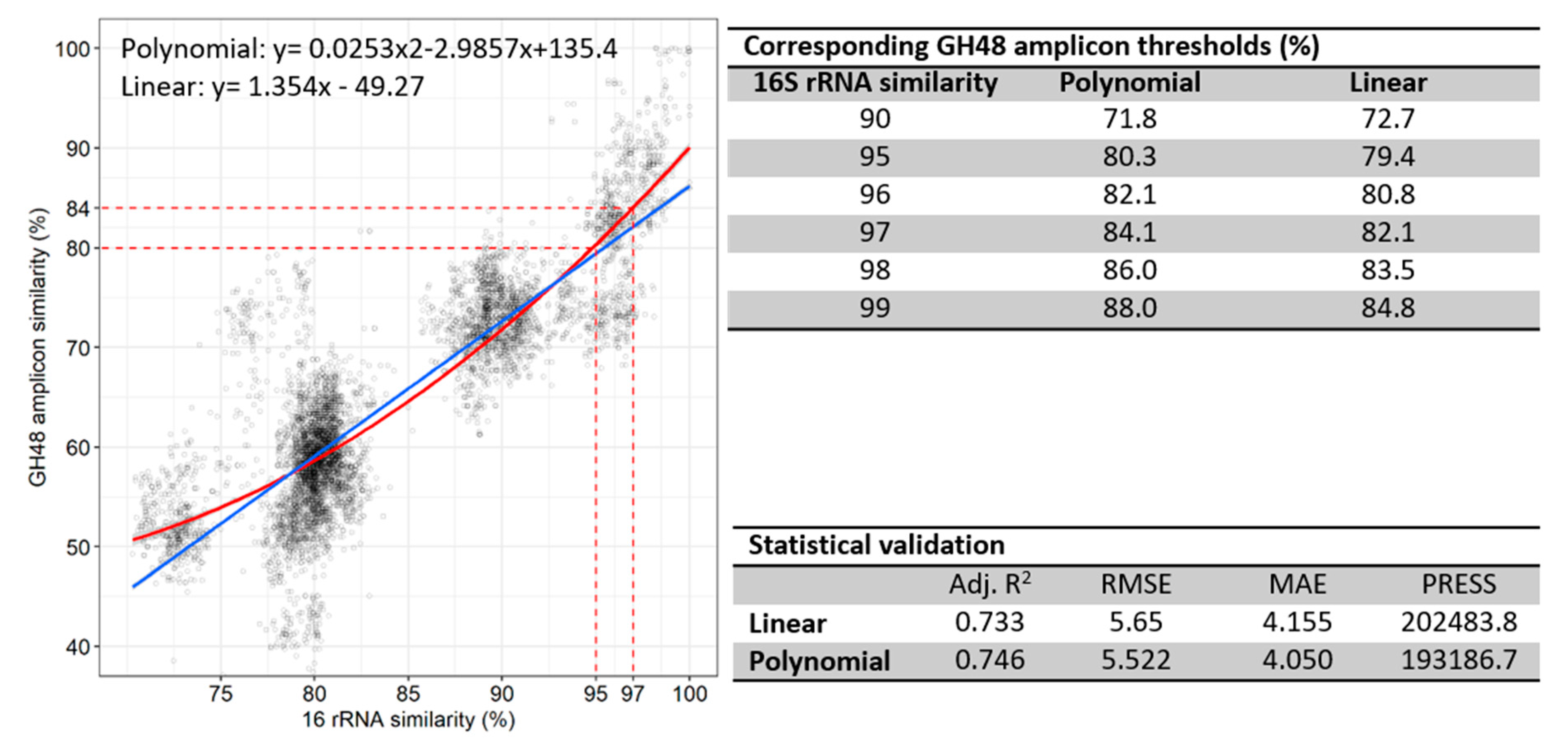
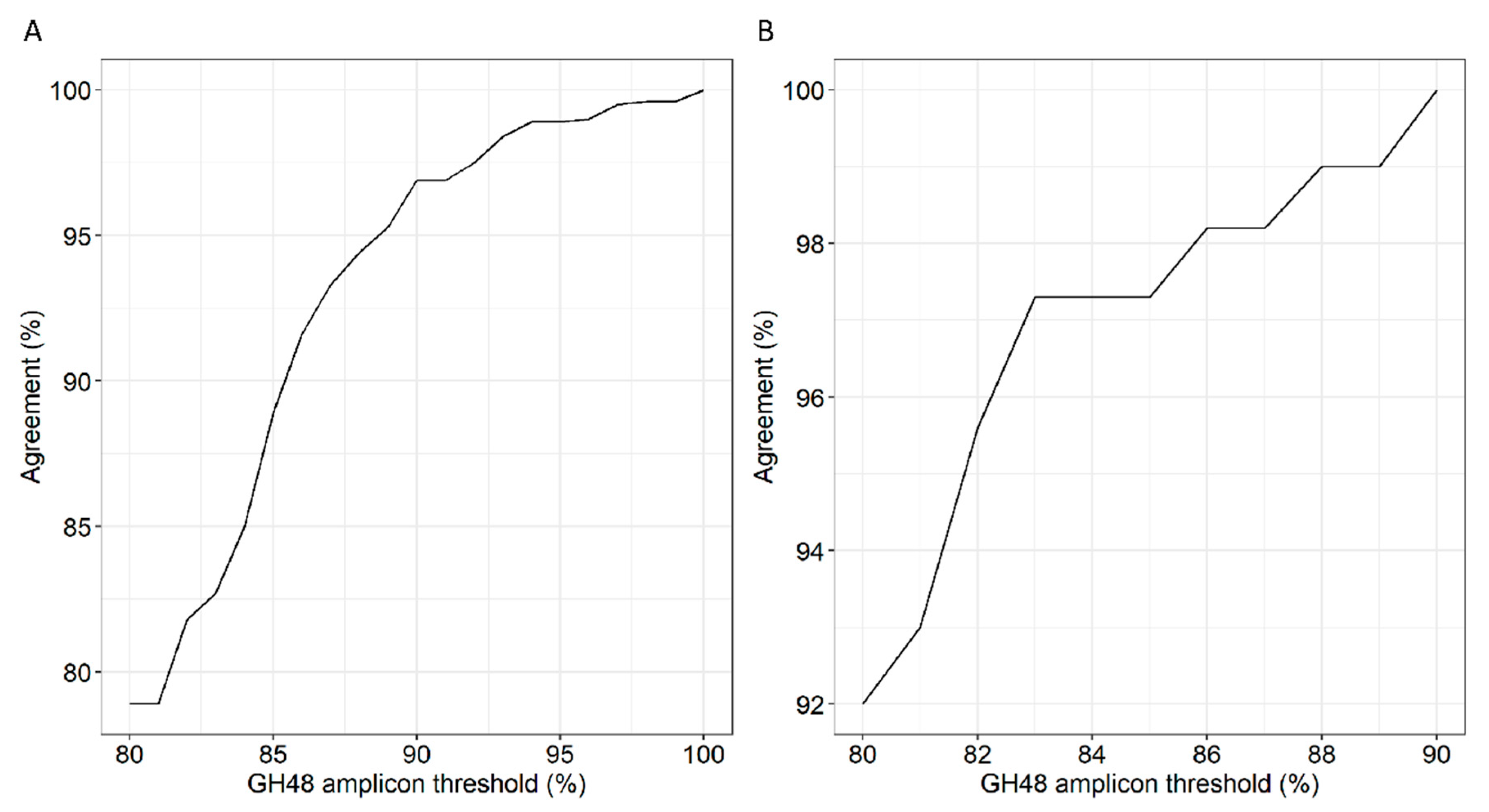
3.5. Correlation of 16S rRNA to GH48 Gene Sequences
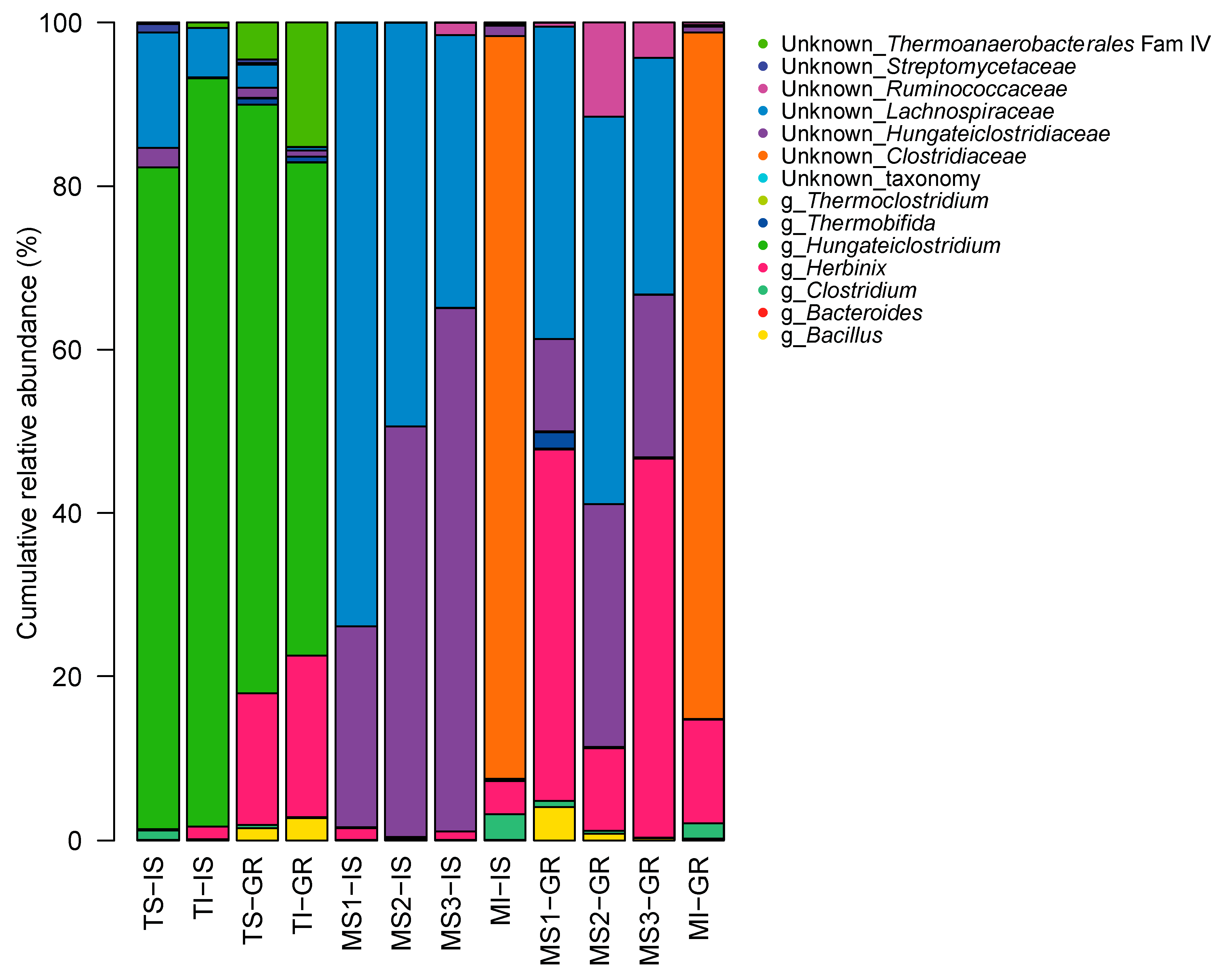
3.6. GH48 Gene Amplicon Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Abbreviations
| CAZy(mes) | carbohydrate-active enzyme(s) |
| ddH2O | doubled deionized water |
| GH48 | glycoside hydrolase of family 48 |
| GR | digestate (from German: Gärrest) |
| IS | in sacco |
| MAE | mean absolute error |
| MI | mesophilic-instable |
| MPN | most probable number |
| MS | mesophilic-stable |
| NGS | next-generation sequencing |
| NTC | non-template control |
| OTU | operational taxonomic unit |
| PRESS | predicted residual error sum of squares |
| qPCR | quantitative polymerase chain reaction |
| RMSE | root-mean-square error |
| RT | reverse transcription |
| TI | thermophilic-instable |
| TS | thermophilic-stable |
| txid | taxonomic identification number |
References
- Naik, S.; Goud, V.V.; Rout, P.K.; Dalai, A.K. Production of first and second generation biofuels: A comprehensive review. Renew. Sustain. Energy Rev. 2010, 14, 578–597. [Google Scholar] [CrossRef]
- Mood, S.H.; Golfeshan, A.H.; Tabatabaei, M.; Jouzani, G.S.; Najafi, G.; Gholami, M.; Ardjmand, M. Lignocellulosic biomass to bioethanol, a comprehensive review with a focus on pretreatment. Renew. Sustain. Energy Rev. 2013, 27, 77–93. [Google Scholar] [CrossRef]
- Zverlov, V.V.; Köck, D.E.; Schwarz, W.H. The role of cellulose-hydrolyzing bacteria in the production of biogas from plant biomass. In Microorganisms in Biorefineries; Kamm, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 335–361. ISBN 978-3-662-45208-0. [Google Scholar]
- Antoni, D.; Zverlov, V.V.; Schwarz, W.H. Biofuels from microbes. Appl. Microbiol. Biotechnol. 2007, 77, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, C.; Sosa, F.M.R.; García, B.D. Enzymatic hydrolysis of biomass from wood. Microb. Biotechnol. 2016, 9, 149–156. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Genomic potential for polysaccharide deconstruction in bacteria. Appl. Environ. Microbiol. 2015, 81, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Goebel, B.M.; Pace, N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 1998, 180, 4765–4774. [Google Scholar] [CrossRef]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef]
- Ward, D.M.; Weller, R.; Bateson, M.M. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 1990, 345, 63–65. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Lebuhn, M.; Hanreich, A.; Klocke, M.; Schlüter, A.; Bauer, C.; Pérez, C.M. Towards molecular biomarkers for biogas production from lignocellulose-rich substrates. Anaerobe 2014, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Quince, C.; Pijl, A.S.; De Hollander, M.; Kowalchuk, G.A. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS ONE 2012, 7, e30600. [Google Scholar] [CrossRef] [PubMed]
- Pereyra, L.P.; Hiibel, S.R.; Riquelme, M.V.P.; Reardon, K.F.; Pruden, A. Detection and quantification of functional genes of cellulose-degrading, fermentative, and sulfate-reducing bacteria and methanogenic archaea. Appl. Environ. Microbiol. 2010, 76, 2192–2202. [Google Scholar] [CrossRef] [PubMed]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar] [CrossRef]
- Izquierdo, J.A.; Sizova, M.V.; Lynd, L.R. Diversity of bacteria and glycosyl hydrolase family 48 genes in cellulolytic consortia enriched from thermophilic biocompost. Appl. Environ. Microbiol. 2010, 76, 3545–3553. [Google Scholar] [CrossRef] [PubMed]
- E Koeck, D.; Pechtl, A.; Zverlov, V.V.; Schwarz, W.H. Genomics of cellulolytic bacteria. Curr. Opin. Biotechnol. 2014, 29, 171–183. [Google Scholar] [CrossRef]
- Zverlov, V.V.; Hiegl, W.; Köck, D.E.; Kellermann, J.; Köllmeier, T.; Schwarz, W.H. Hydrolytic bacteria in mesophilic and thermophilic degradation of plant biomass. Eng. Life Sci. 2010, 10, 528–536. [Google Scholar] [CrossRef]
- Dai, X.; Tian, Y.; Li, J.; Su, X.; Wang, X.; Zhao, S.; Liu, L.; Luo, Y.; Liu, D.; Zheng, H.; et al. Metatranscriptomic analyses of plant cell wall polysaccharide degradation by microorganisms in the cow rumen. Appl. Environ. Microbiol. 2015, 81, 1375–1386. [Google Scholar] [CrossRef]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280 Pt 2, 309–316. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, X.; Tian, Y.; Dong, Z.; Hu, S.; Huang, L.; Dai, X. Gene diversity of the bacterial 48 family glycoside hydrolase (GH48) in rumen environment. Wei Sheng Wu Xue Bao = Acta Microbiol. Sin. 2014, 54, 53–61. [Google Scholar]
- Maus, I.; Koeck, D.E.; Cibis, K.G.; Hahnke, S.; Kim, Y.S.; Langer, T.; Kreubel, J.; Erhard, M.; Bremges, A.; Off, S.; et al. Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol. Biofuels 2016, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J. The next frontier of the anaerobic digestion microbiome: From ecology to process control. Environ. Sci. Ecotechnol. 2020, 3, 100032. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Boil. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Boil. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Rudi, K.; Skulberg, O.M.; Larsen, F.; Jakobsen, K.S. Strain characterization and classification of oxyphotobacteria in clone cultures on the basis of 16S rRNA sequences from the variable regions V6, V7, and V8. Appl. Environ. Microbiol. 1997, 63, 2593–2599. [Google Scholar] [CrossRef]
- Costa, R.; Götz, M.; Mrotzek, N.; Lottmann, J.; Berg, G.; Smalla, K. Effects of site and plant species on rhizosphere community structure as revealed by molecular analysis of microbial guilds. FEMS Microbiol. Ecol. 2006, 56, 236–249. [Google Scholar] [CrossRef]
- Mohamed, R.; Chaudhry, A.S. Methods to study degradation of ruminant feeds. Nutr. Res. Rev. 2008, 21, 68–81. [Google Scholar] [CrossRef]
- Koeck, D.E.; Mechelke, M.; Zverlov, V.V.; Liebl, W.; Schwarz, W.H. Herbivorax saccincola gen. nov., sp. nov., a cellulolytic, anaerobic, thermophilic bacterium isolated via in sacco enrichments from a lab-scale biogas reactor. Int. J. Syst. Evol. Microbiol. 2016, 66, 4458–4463. [Google Scholar] [CrossRef]
- Rettenmaier, R.; Schneider, M.; Munk, B.; Lebuhn, M.; Jünemann, S.; Sczyrba, A.; Maus, I.; Zverlov, V.V.; Liebl, W. Importance of Defluviitalea raffinosedens for hydrolytic biomass degradation in co-culture with Hungateiclostridium thermocellum. Microorganisms 2020, 8, 915. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2009, 37, D5–D15. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Edgar, R.C. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 2018, 34, 2371–2375. [Google Scholar] [CrossRef]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B.A. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Genet. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Berry, D.; Ben Mahfoudh, K.; Wagner, M.; Loy, A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 2011, 77, 7846–7849. [Google Scholar] [CrossRef]
- Reitmeier, S.; Kiessling, S.; Clavel, T.; List, M.; Almeida, E.L.; Ghosh, T.; Neuhaus, K.; Grallert, H.; Troll, M.; Rathmann, W.; et al. Arrhythmic gut microbiome signatures for risk profiling of Type-2 diabetes. SSRN Electron. J. 2019. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Tindall, B.J.; Busse, H.-J.; Ludwig, W.; Rosselló-Mora, R.; Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal Gene Transfer in Prokaryotes: Quantification and Classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef]
- Sukharnikov, L.O.; Alahuhta, M.; Brunecky, R.; Upadhyay, A.; Himmel, M.E.; Lunin, V.V.; Zhulin, I.B. Sequence, structure, and evolution of cellulases in glycoside hydrolase family 48. J. Boil. Chem. 2012, 287, 41068–41077. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, C.; Abu Al-Soud, W.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef]
- Rui, J.; Li, J.; Zhang, S.; Yan, X.; Wang, Y.; Li, X. The core populations and co-occurrence patterns of prokaryotic communities in household biogas digesters. Biotechnol. Biofuels 2015, 8, 1–15. [Google Scholar] [CrossRef]
- Hua, M.; Yu, S.; Ma, Y.; Chen, S.; Li, F. Genetic diversity detection and gene discovery of novel glycoside hydrolase family 48 from soil environmental genomic DNA. Ann. Microbiol. 2018, 68, 163–174. [Google Scholar] [CrossRef]
- Maus, I.; Bremges, A.; Stolze, Y.; Hahnke, S.; Cibis, K.G.; Koeck, D.E.; Kim, Y.S.; Kreubel, J.; Hassa, J.; Wibberg, D.; et al. Genomics and prevalence of bacterial and archaeal isolates from biogas-producing microbiomes. Biotechnol. Biofuels 2017, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Koeck, D.E.; Ludwig, W.; Wanner, G.; Zverlov, V.V.; Liebl, W.; Schwarz, W.H. Herbinix hemicellulosilytica gen. nov., sp. nov., a thermophilic cellulose-degrading bacterium isolated from a thermophilic biogas reactor. Int. J. Syst. Evol. Microbiol. 2015, 65, 2365–2371. [Google Scholar] [CrossRef] [PubMed]
- Koeck, D.E.; Hahnke, S.; Zverlov, V.V. Herbinix luporum sp. nov., a thermophilic cellulose-degrading bacterium isolated from a thermophilic biogas reactor. Int. J. Syst. Evol. Microbiol. 2016, 66, 4132–4137. [Google Scholar] [CrossRef]
- Mechelke, M.; Koeck, D.; Broeker, J.; Roessler, B.; Krabichler, F.; Schwarz, W.H.; Zverlov, V.V.; Liebl, W. Characterization of the arabinoxylan-degrading machinery of the thermophilic bacterium Herbinix hemicellulosilytica—Six new xylanases, three arabinofuranosidases and one xylosidase. J. Biotechnol. 2017, 257, 122–130. [Google Scholar] [CrossRef]
- Rettenmaier, R.; Gerbaulet, M.; Liebl, W.; Zverlov, V.V. Hungateiclostridium mesophilum sp. nov., a mesophilic, cellulolytic and spore-forming bacterium isolated from a biogas fermenter fed with maize silage. Int. J. Syst. Evol. Microbiol. 2019, 69, 3567–3573. [Google Scholar] [CrossRef]
- Lebuhn, M.; Effenberger, M.; Gronauer, A.; Wilderer, P.; Wuertz, S. Using quantitative real-time PCR to determine the hygienic status of cattle manure. Water Sci. Technol. 2003, 48, 97–103. [Google Scholar] [CrossRef]
- Rettenmaier, R.; Duerr, C.; Neuhaus, K.; Liebl, W.; Zverlov, V.V. Comparison of sampling techniques and different media for the enrichment and isolation of cellulolytic organisms from biogas fermenters. Syst. Appl. Microbiol. 2019, 42, 481–487. [Google Scholar] [CrossRef]
- Orhorhoro, E.K.; Ebunilo, P.O.; Sadjere, G.E. Effect of organic loading rate (OLR) on biogas yield using a single and three-stages continuous anaerobic digestion reactors. Int. J. Eng. Res. Afr. 2018, 39, 147–155. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primer-ID | Target Gene | Nucleotide Sequence | Source |
|---|---|---|---|
| Cel48_490F_I | GH48 | T[I]ATGGTTGAAGCTCCDGAYTAYGG | Pereyra et al. 2010 [14], modified within this work |
| Cel48_920R_I | GH48 | CCAAA[I]CCRTACCAGTTRTCAACRTC | |
| F919mod | bacterial 16S rRNA | GAATTGACGGGGRYCCGCACAAG | Rudi et al. 1997 [28] modified by Lebuhn et al. 2014 [12] |
| R1378mod | bacterial 16S rRNA | CGGTGTGTACAAGRCCCGRGAACG | Costa et al. 2006 [29], modified by Lebuhn et al. 2014 [12] |
| Cel48-Mix2F | GH48 | Table 2 | this work |
| Cel48-Mix2R | GH48 | Table 2 |
| ID | Sequence | Length (bp) | V (µL) | N (Degeneration) |
|---|---|---|---|---|
| F1 | TCTTGAGTGAAGCTCCAGACTA | 22 | 1 | 0 |
| F2 | TGCTTAGCGA[AG]GCGCCCAA | 19 | 2 | 1 |
| F3 | GAGCGA[AG]GC[I]CC[I]GATTAC | 19 | 2 | 1 |
| F4 | TGAGCGAGGCTCC[GT]GAC[CT]A | 19 | 4 | 2 |
| F5 | TGATATGCGAAGCACCTGA[CT]TATG | 24 | 2 | 1 |
| F6 | TCATATG[CT]GAAGC[GA]CC[TG]GATTAC | 23 | 8 | 3 |
| F7 | TGATATGCGAGGCACCGGATTA | 22 | 1 | 0 |
| F8 | TGAACTGTGAAGCTCCTGATCAA | 23 | 1 | 0 |
| F9 | TGTGTGTTGAAGCGCCTGATTAC | 23 | 1 | 0 |
| F10 | TGATTGT[I]GAAGCTCC[TG]GA[CT]TATG | 24 | 4 | 2 |
| F11 | ATCGTCGAAGCGCCTGACCA | 20 | 1 | 0 |
| F12 | ATCGACGAGGCGCCCGA | 17 | 1 | 0 |
| F13 | TTTTGGTGGAAGCTCCGGACTAT | 23 | 1 | 0 |
| F14 | TAATGGTTGAAGCACC[I]GAC[CT]AT | 23 | 2 | 1 |
| F15 | TCATCGTCGA[GA]GC[I]CC[I]GA[CT]TA | 22 | 4 | 2 |
| F16n | ATCGTCGAGGC[I]CC[GC]GACC | 19 | 2 | 1 |
| R1 | CCGAAGCCGTAGACGTTGTC | 20 | 1 | 0 |
| R2 | CCGTAACCGTAGATGTTGTC[I]A | 22 | 1 | 0 |
| R3 | CCG[AT]AGCCGTA[GC][GC]TGTTGTC | 20 | 8 | 3 |
| R4 | CCGTATCCATACCAGTTATCCGTA | 24 | 1 | 0 |
| R5 | CCAAATCCATACCAGTTATCACAGT | 25 | 1 | 0 |
| R6 | CCAAAACCATACCAGTTATCAACATC | 26 | 1 | 0 |
| R7 | CCATCCATACCAGTT[AG]TC[I]AC[AG]TC | 24 | 4 | 2 |
| R8 | CCR[AT]A[I]CCGTAGACGTTGTC | 20 | 4 | 1 |
| R9 | CCGAAGCCGTACCA[AG]TT[AG]TC | 20 | 4 | 2 |
| Sample | GH48 (Gene Copies/µL) | Standard Deviation | 16S rRNA (Gene Copies/µL) | Standard Deviation | Ratio GH48/16S rRNA (%) |
|---|---|---|---|---|---|
| MS1 | 5.7 × 106 | ±1.1 × 106 | 7.1 × 108 | ± 1.8 × 108 | 0.8 |
| MS2 | 4.8 × 106 | ±3.1 × 105 | 1.5 × 109 | ± 3.6 × 108 | 0.3 |
| MS3 | 3.6 × 106 | ±4.7 × 105 | 1.2 × 109 | ± 1.5 × 108 | 0.3 |
| MI | 5.4 × 106 | ±4.0 × 105 | 6.7 × 108 | ± 1.5 × 107 | 0.8 |
| TS | 2.9 × 106 | ±2.9 × 105 | 8.0 × 108 | ± 2.6 × 108 | 0.4 |
| TI | 7.9 × 106 | ±8.1 × 105 | 1.2 × 109 | ± 6.8 × 107 | 0.7 |
| Sample | GH48 (Gene Copies/µL) | Standard Deviation | 16S rRNA (Gene Copies/µL) | Standard Deviation | Ratio GH48/16S rRNA (%) |
|---|---|---|---|---|---|
| MS1 | 6.8 × 106 | ±8.7 × 105 | 2.2 × 108 | ±5.1 × 107 | 3.1 |
| MS2 | 2.4 × 107 | ±9.6 × 106 | 6.4 × 108 | ±1.1 × 107 | 3.7 |
| MS3 | 3.8 × 106 | ±1.4 × 105 | 1.9 × 108 | ±2.0 × 107 | 2.0 |
| MI | 9.1 × 104 | ±1.3 × 104 | 7.8 × 106 | ±8.4 × 105 | 1.2 |
| TS | 3.4 × 106 | ±4.0 × 105 | 2.4 × 108 | ±4.1 × 107 | 1.4 |
| TI | 5.8 × 106 | ±1.5 × 106 | 1.7 × 108 | ±1.4 × 107 | 3.5 |
| Entries Summary | Sequence Length Distribution | |||||
|---|---|---|---|---|---|---|
| No. of entries | 944 | Min | Mean | Max | ||
| No. of genera | 104 | GH48 module containing protein (nucleotide) | 1429 | 2926 | 7032 | |
| No. of families | 42 | GH48 module containing protein (amino acid) | 475 | 975 | 2343 | |
| Entries with full-length 16S | 580 (61%) | GH48 amplicon length (nucleotide, without primers) | 291 | 344 | 379 | |
| Species or strains with >1 GH48 | 82 (8.7%) | 16S rRNA gene sequence (nucleotide) | 72 | 1459 | 1834 | |
| Entries with identical GH48 module | 78 (8.3%) | |||||
| Entries with identical GH48 amplicon | 184 (19.5%) | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rettenmaier, R.; Lo, Y.K.; Schmidt, L.; Munk, B.; Lagkouvardos, I.; Neuhaus, K.; Schwarz, W.; Liebl, W.; Zverlov, V. A Novel Primer Mixture for GH48 Genes: Quantification and Identification of Truly Cellulolytic Bacteria in Biogas Fermenters. Microorganisms 2020, 8, 1297. https://doi.org/10.3390/microorganisms8091297
Rettenmaier R, Lo YK, Schmidt L, Munk B, Lagkouvardos I, Neuhaus K, Schwarz W, Liebl W, Zverlov V. A Novel Primer Mixture for GH48 Genes: Quantification and Identification of Truly Cellulolytic Bacteria in Biogas Fermenters. Microorganisms. 2020; 8(9):1297. https://doi.org/10.3390/microorganisms8091297
Chicago/Turabian StyleRettenmaier, Regina, Yat Kei Lo, Larissa Schmidt, Bernhard Munk, Ilias Lagkouvardos, Klaus Neuhaus, Wolfgang Schwarz, Wolfgang Liebl, and Vladimir Zverlov. 2020. "A Novel Primer Mixture for GH48 Genes: Quantification and Identification of Truly Cellulolytic Bacteria in Biogas Fermenters" Microorganisms 8, no. 9: 1297. https://doi.org/10.3390/microorganisms8091297
APA StyleRettenmaier, R., Lo, Y. K., Schmidt, L., Munk, B., Lagkouvardos, I., Neuhaus, K., Schwarz, W., Liebl, W., & Zverlov, V. (2020). A Novel Primer Mixture for GH48 Genes: Quantification and Identification of Truly Cellulolytic Bacteria in Biogas Fermenters. Microorganisms, 8(9), 1297. https://doi.org/10.3390/microorganisms8091297