Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Sequencing
2.2. Assembly and Binning
2.3. Relative Abundance, Functional Characterization and Metabolic Analyses
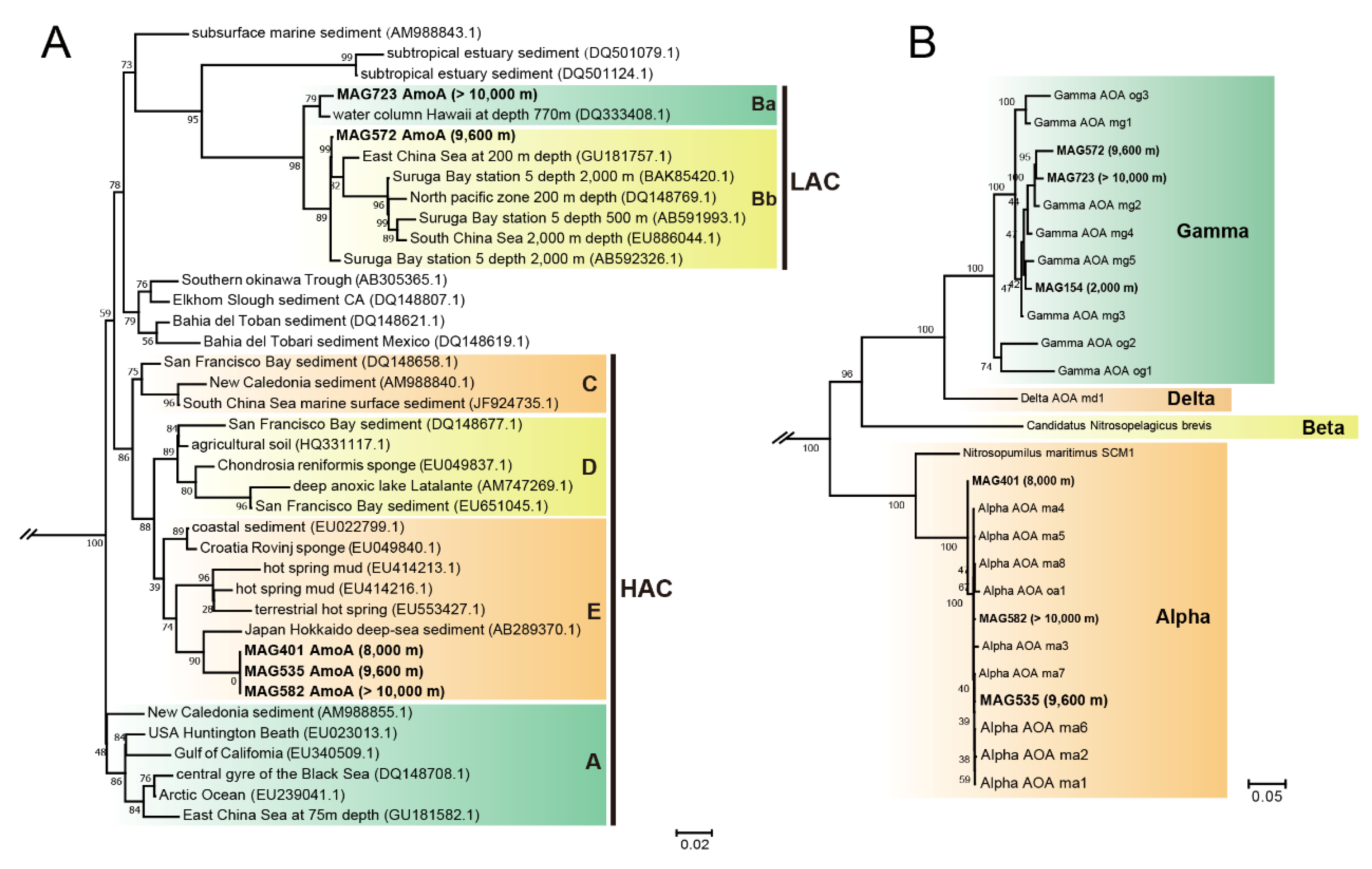
2.4. Phylogenetic Analyses of the Reconstructed Genomes, AmoA Proteins and AOA Genomes
2.5. Data Availability
3. Results
3.1. Phylogenetic Diversity through the Water Column in the Mariana Trench
3.2. Phototrophy and Carbon Fixation
3.3. Complex Carbon Degradation
3.4. Central Metabolism
3.5. Fermentative Metabolism
3.6. Nitrogen Cycling
3.7. Sulfur Cycling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nunoura, T.; Takaki, Y.; Hirai, M.; Shimamura, S.; Makabe, A.; Koide, O.; Kikuchi, T.; Miyazaki, J.; Koba, K.; Yoshida, N. Hadal biosphere: Insight into the microbial ecosystem in the deepest ocean on Earth. Proc. Natl. Acad. Sci. USA 2015, 112, E1230–E1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zheng, Y.; Lin, H.; Wang, X.; Li, M.; Liu, Y.; Yu, M.; Zhao, M.; Pedentchouk, N.; Lea-Smith, D.J. Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana Trench. Microbiome 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wang, L.; Liu, Q.; Wang, Z.; Li, Z.; Fang, J.; Zhang, L.; Luo, M. Depth-resolved distribution of particle-attached and free-living bacterial communities in the water column of the New Britain Trench. Front. Microbiol. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkowski, P.G. The role of phytoplankton photosynthesis in global biogeochemical cycles. Photosynth. Res. 1994, 39, 235–258. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Consarnau, L.; Raven, J.A.; Levine, N.M.; Cutter, L.S.; Wang, D.; Seegers, B.; Arístegui, J.; Fuhrman, J.A.; Gasol, J.M.; Sañudo-Wilhelmy, S.A. Microbial rhodopsins are major contributors to the solar energy captured in the sea. Sci. Adv. 2019, 5, eaaw8855. [Google Scholar] [CrossRef] [Green Version]
- Hansell, D.; Carlson, C.; Repeta, D.; Schlitzer, R. Dissolved organic matter in the ocean: New insights stimulated by a controversy. Oceanography 2009, 22, 202–211. [Google Scholar] [CrossRef]
- Könneke, M.; Schubert, D.M.; Brown, P.C.; Hügler, M.; Standfest, S.; Schwander, T.; von Borzyskowski, L.S.; Erb, T.J.; Stahl, D.A.; Berg, I.A. Ammonia-oxidizing archaea use the most energy-efficient aerobic pathway for CO2 fixation. Proc. Natl. Acad. Sci. USA 2014, 111, 8239–8244. [Google Scholar] [CrossRef] [Green Version]
- Massana, R.; Murray, A.E.; Preston, C.M.; DeLong, E.F. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl. Environ. Microbiol. 1997, 63, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Massana, R.; DeLong, E.F.; Pedrós-Alió, C. A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces. Appl. Environ. Microbiol. 2000, 66, 1777–1787. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; Xie, W.; Martin-Cuadrado, A.-B.; Rodriguez-Valera, F. Marine Group II Archaea, potentially important players in the global ocean carbon cycle. Front. Microbiol. 2015, 6, 1108. [Google Scholar] [CrossRef] [Green Version]
- Nunoura, T.; Hirai, M.; Yoshida-Takashima, Y.; Nishizawa, M.; Kawagucci, S.; Yokokawa, T.; Miyazaki, J.; Koide, O.; Makita, H.; Takaki, Y. Distribution and niche separation of planktonic microbial communities in the water columns from the surface to the hadal waters of the Japan Trench under the Eutrophic Ocean. Front. Microbiol. 2016, 7, 1261. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, G.; Cornejo-Castillo, F.M.; Benítez-Barrios, V.; Fraile-Nuez, E.; Álvarez-Salgado, X.A.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 2016, 10, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, A.J.; Fujii, T.; Mayor, D.J.; Solan, M.; Priede, I.G. Hadal trenches: The ecology of the deepest places on Earth. Trends Ecol. Evol. 2010, 25, 190–197. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.M.; Cui, G.J.; Nunoura, T.; Takaki, Y.; Li, W.L.; Li, J.; Gao, Z.M.; Takai, K.; Zhang, A.Q. Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ. Microbiol. 2019, 21, 716–729. [Google Scholar] [CrossRef]
- Zhong, H.; Lehtovirta-Morley, L.; Liu, J.; Zheng, Y.; Lin, H.; Song, D.; Todd, J.; Tian, J.; Zhang, X. Novel insights into the Thaumarchaeota in the deepest oceans: Their metabolism and potential adaptation mechanisms. Microbiome 2020, 8, 78. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1. 0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Kingsford, C. Salmon: Accurate, versatile and ultrafast quantification from RNA-seq data using lightweight-alignment. Biorxiv 2015, 021592. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. Available online: https://www.osti.gov/servlets/purl/1241166 (accessed on 5 June 2019).
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xia, R.; Chen, H.; He, Y. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. BioRxiv 2018, 289660. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.-I.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Baltar, F.; Arístegui, J.; Sintes, E.; Van Aken, H.M.; Gasol, J.M.; Herndl, G.J. Prokaryotic extracellular enzymatic activity in relation to biomass production and respiration in the meso-and bathypelagic waters of the (sub) tropical Atlantic. Environ. Microbiol. 2009, 11, 1998–2014. [Google Scholar] [CrossRef]
- Arnosti, C. Patterns of microbially driven carbon cycling in the ocean: Links between extracellular enzymes and microbial communities. Adv. Oceanogr. 2014, 2014, 706082. [Google Scholar] [CrossRef] [Green Version]
- Haro-Moreno, J.M.; López-Pérez, M.; José, R.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Fine metagenomic profile of the Mediterranean stratified and mixed water columns revealed by assembly and recruitment. Microbiome 2018, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Wolf, Y.I. Genomics of bacteria and archaea: The emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008, 36, 6688–6719. [Google Scholar] [CrossRef] [Green Version]
- Biller, S.J.; Berube, P.M.; Lindell, D.; Chisholm, S.W. Prochlorococcus: The structure and function of collective diversity. Nature Rev. Microbiol. 2015, 13, 13–27. [Google Scholar] [CrossRef]
- Bendall, M.L.; Stevens, S.L.; Chan, L.-K.; Malfatti, S.; Schwientek, P.; Tremblay, J.; Schackwitz, W.; Martin, J.; Pati, A.; Bushnell, B. Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. ISME J. 2016, 10, 1589–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya, J.P.; Holl, C.M.; Zehr, J.P.; Hansen, A.; Villareal, T.A.; Capone, D.G. High rates of N2 fixation by unicellular diazotrophs in the oligotrophic Pacific Ocean. Nature 2004, 430, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.; Swan, B.K.; Poulton, N.J.; Gomez, M.L.; Masland, D.; Sieracki, M.E.; Stepanauskas, R. High-throughput single-cell sequencing identifies photoheterotrophs and chemoautotrophs in freshwater bacterioplankton. ISME J. 2012, 6, 113–123. [Google Scholar] [CrossRef] [Green Version]
- He, X.; McLean, J.S.; Edlund, A.; Yooseph, S.; Hall, A.P.; Liu, S.-Y.; Dorrestein, P.C.; Esquenazi, E.; Hunter, R.C.; Cheng, G. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. USA 2015, 112, 244–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücker, S.; Wagner, M.; Maixner, F.; Pelletier, E.; Koch, H.; Vacherie, B.; Rattei, T.; Damsté, J.S.S.; Spieck, E.; Le Paslier, D. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 13479–13484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, P.; Hahnke, R.L.; Unfried, F.; Markert, S.; Huang, S.; Barbeyron, T.; Harder, J.; Becher, D.; Schweder, T.; Glöckner, F.O. Niches of two polysaccharide-degrading Polaribacter isolates from the North Sea during a spring diatom bloom. ISME J. 2015, 9, 1410–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennke, C.M.; Krüger, K.; Kappelmann, L.; Huang, S.; Gobet, A.; Schüler, M.; Barbe, V.; Fuchs, B.M.; Michel, G.; Teeling, H. Polysaccharide utilisation loci of Bacteroidetes from two contrasting open ocean sites in the North Atlantic. Environ. Microbiol. 2016, 18, 4456–4470. [Google Scholar] [CrossRef]
- Liu, J.; Xue, C.-X.; Sun, H.; Zheng, Y.; Meng, Z.; Zhang, X.-H. Carbohydrate catabolic capability of a Flavobacteriia bacterium isolated from hadal water. Syst. Appl. Microbiol. 2019, 42, 263–274. [Google Scholar] [CrossRef]
- Xue, C.X.; Zhang, H.; Lin, H.Y.; Sun, Y.; Luo, D.; Huang, Y.; Zhang, X.H.; Luo, H. Ancestral niche separation and evolutionary rate differentiation between sister marine Flavobacteria lineages. Environ. Microbiol. 2020, 22, 3234–3247. [Google Scholar] [CrossRef]
- Preisig, O.; Zufferey, R.; Thöny-Meyer, L.; Appleby, C.A.; Hennecke, H. A high-affinity cbb3-type cytochrome oxidase terminates the symbiosis-specific respiratory chain of Bradyrhizobium japonicum. J. Bacteriol. 1996, 178, 1532–1538. [Google Scholar] [CrossRef] [Green Version]
- Lam, P.; Kuypers, M.M. Microbial nitrogen cycling processes in oxygen minimum zones. Ann. Rev. Mar. Sci. 2011, 3, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Kessler, A.J.; Grace, M.R.; Cook, P.L. Increased rates of dissimilatory nitrate reduction to ammonium (DNRA) under oxic conditions in a periodically hypoxic estuary. Geochim. Cosmochim. Acta 2014, 133, 313–324. [Google Scholar] [CrossRef]
- Friedrich, C.G.; Rother, D.; Bardischewsky, F.; Quentmeier, A.; Fischer, J. Oxidation of reduced inorganic sulfur compounds by bacteria: Emergence of a common mechanism? Appl. Environ. Microbiol. 2001, 67, 2873–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigaard, N.-U.; Dahl, C. Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 2008, 54, 103–200. [Google Scholar]
- Bamford, V.A.; Bruno, S.; Rasmussen, T.; Appia-Ayme, C.; Cheesman, M.R.; Berks, B.C.; Hemmings, A.M. Structural basis for the oxidation of thiosulfate by a sulfur cycle enzyme. EMBO J. 2002, 21, 5599–5610. [Google Scholar] [CrossRef]
- Bartlett, D.H. Microbial life in the trenches. Mar. Technol. Soc. J. 2009, 43, 128–131. [Google Scholar] [CrossRef]
- Ramirez-Llodra, E.; Brandt, A.; Danovaro, R.; De Mol, B.; Escobar, E.; German, C.; Levin, L.; Arbizu, P.; Menot, L.; Buhl-Mortensen, P. Deep, diverse and definitely different: Unique attributes of the world’s largest ecosystem. Biogeosciences 2010, 7, 2851–2899. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, S.L.; Hannisdal, B.; Lanzén, A.; Baumberger, T.; Flesland, K.; Fonseca, R.; Øvreås, L.; Steen, I.H.; Thorseth, I.H.; Pedersen, R.B. Correlating microbial community profiles with geochemical data in highly stratified sediments from the Arctic Mid-Ocean Ridge. Proc. Natl. Acad. Sci. USA 2012, 109, E2846–E2855. [Google Scholar] [CrossRef] [Green Version]
- Danovaro, R.; Snelgrove, P.V.; Tyler, P. Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 2014, 29, 465–475. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Zhou, S.; Zheng, Y.; Wu, Y.; Kogure, K.; Zhang, X.-H. Diversity of culturable heterotrophic bacteria from the Mariana Trench and their ability to degrade macromolecules. Mar. Life Sci. Technol. 2020, 2, 181–193. [Google Scholar] [CrossRef] [Green Version]
- van Oijen, T.; van Leeuwe, M.A.; Gieskes, W.W. Variation of particulate carbohydrate pools over time and depth in a diatom-dominated plankton community at the Antarctic Polar Front. Polar Biol. 2003, 26, 195–201. [Google Scholar] [CrossRef]
- Li, W.-L.; Huang, J.-M.; Zhang, P.-W.; Cui, G.-J.; Wei, Z.-F.; Wu, Y.-Z.; Gao, Z.-M.; Han, Z.; Wang, Y. Periodic and spatial spreading of alkanes and Alcanivorax bacteria in deep waters of the Mariana Trench. Appl. Environ. Microbiol. 2019, 85, e02089-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glud, R.N.; Wenzhöfer, F.; Middelboe, M.; Oguri, K.; Turnewitsch, R.; Canfield, D.E.; Kitazato, H. High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth. Nat. Geosci. 2013, 6, 284–288. [Google Scholar] [CrossRef]
- Carvalho, V.M.L. Metagenomic Analysis of Mariana Trench Sediment Samples. Master’s Thesis, University of Copenhagen, København, Denmark, 2013. [Google Scholar]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. BBA-Bioenerg. 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, C.-X.; Liu, J.; Lea-Smith, D.J.; Rowley, G.; Lin, H.; Zheng, Y.; Zhu, X.-Y.; Liang, J.; Ahmad, W.; Todd, J.D.; et al. Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth. Microorganisms 2020, 8, 1309. https://doi.org/10.3390/microorganisms8091309
Xue C-X, Liu J, Lea-Smith DJ, Rowley G, Lin H, Zheng Y, Zhu X-Y, Liang J, Ahmad W, Todd JD, et al. Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth. Microorganisms. 2020; 8(9):1309. https://doi.org/10.3390/microorganisms8091309
Chicago/Turabian StyleXue, Chun-Xu, Jiwen Liu, David J. Lea-Smith, Gary Rowley, Heyu Lin, Yanfen Zheng, Xiao-Yu Zhu, Jinchang Liang, Waqar Ahmad, Jonathan D. Todd, and et al. 2020. "Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth" Microorganisms 8, no. 9: 1309. https://doi.org/10.3390/microorganisms8091309
APA StyleXue, C.-X., Liu, J., Lea-Smith, D. J., Rowley, G., Lin, H., Zheng, Y., Zhu, X.-Y., Liang, J., Ahmad, W., Todd, J. D., & Zhang, X.-H. (2020). Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth. Microorganisms, 8(9), 1309. https://doi.org/10.3390/microorganisms8091309