Structural, Functional, and Metabolic Alterations in Human Cerebrovascular Endothelial Cells during Toxoplasma gondii Infection and Amelioration by Verapamil In Vitro

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Maintenance of Cell Cultures
2.2. Propagation and Purification of Toxoplasma gondii Tachyzoites
2.3. Infection of Brain Microvascular Endothelial Cells (BMECs)
2.4. Phenotypic Characterization of Infection
2.4.1. AlamarBlue® Viability Assay
2.4.2. Fluorescence Staining
2.4.3. Scanning Electron Microscopy (SEM)
2.4.4. Transmission Electron Microscopy (TEM)
2.5. Analysis of Neutral Lipids
2.6. Endothelial Cell Integrity and Permeability Studies
2.6.1. TEER Measurements
2.6.2. Fluorescein Isothiocyanate (FITC)-Dextran Leakage through the BMEC Barrier
2.6.3. Evaluation of Parasite-Crossing Using Carboxyfluorescein Succinimidyl Ester (CFSE) Staining
2.6.4. Effect of Infection on the Integrity of the BMEC Monolayer
2.7. 1H NMR-Based Metabolomics
2.7.1. Sample Preparation and Data Acquisition
2.7.2. Spectra Processing and Multivariate Analysis
2.8. The Effect of Verapamil on BMEC Function
2.8.1. Inhibitory Effect of Verapamil on T. gondii Growth
2.8.2. Effect of Verapamil on BMEC Monolayer Integrity
2.9. Statistical Analysis
3. Results
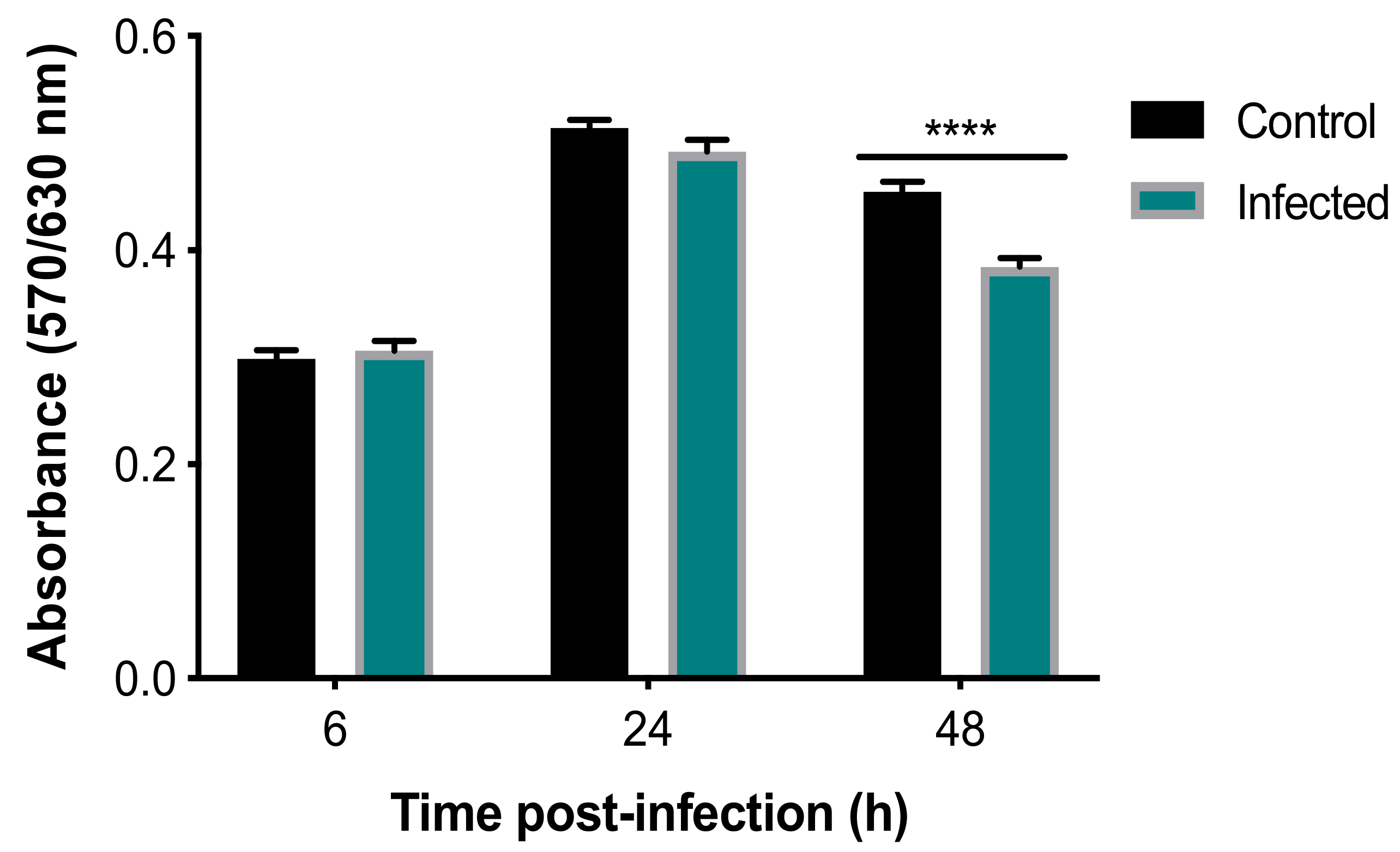
3.1. Effects of T. gondii on the Viability and Structure of BMECs
3.2. Perturbed Lipid Homeostasis in Infected Cells
3.3. Disruption of BME Barrier Integrity by Infection
3.4. Determination of Metabolic Differences between Infected and Uninfected Cells
3.5. Verapamil Inhibits T. gondii Growth and Reduces Barrier Permeability in Infected Cells
4. Discussion
4.1. Reduced Viability and Altered Structure of Infected Cells
4.2. Perturbed Lipid Homeostasis in Infected Cells
4.3. Compromised Barrier Integrity and Increased Parasite Crossing
4.4. Infection-Induced Metabolic Alterations
4.5. Verapamil Ameliorated the Impact of T. gondii on Barrier Integrity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Weiss, L.M.; Dubey, J.P. Toxoplasmosis: A history of clinical observations. Int. J. Parasitol. 2009, 39, 895–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steininger, C.; Popow-Kraupp, T.; Laferl, H.; Seiser, A.; Godl, I.; Djamshidian, S.; Puchhammer-Stockl, E. Acute encephalopathy associated with influenza A virus infection. Clin. Infect. Dis. 2003, 36, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carruthers, V.B.; Suzuki, Y. Effects of Toxoplasma gondii infection on the brain. Schizophr. Bull. 2007, 33, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Melzer, T.C.; Cranston, H.J.; Weiss, L.M.; Halonen, S.K. Host cell preference of Toxoplasma gondii cysts in murine brain: A confocal study. J. Neuroparasitol. 2010, 1. [Google Scholar] [CrossRef]
- Daubener, W.; Spors, B.; Hucke, C.; Adam, R.; Stins, M.; Kim, K.S.; Schroten, H. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect. Immun. 2001, 69, 6527–6531. [Google Scholar] [CrossRef] [Green Version]
- Lambert, H.; Hitziger, N.; Dellacasa, I.; Svensson, M.; Barragan, A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell. Microbiol. 2006, 8, 1611–1623. [Google Scholar] [CrossRef]
- Elsheikha, H.M.; Khan, N.A. Protozoa traversal of the blood-brain barrier to invade the central nervous system. FEMS Microbiol. Rev. 2010, 34, 532–553. [Google Scholar] [CrossRef]
- Lachenmaier, S.M.; Deli, M.A.; Meissner, M.; Liesenfeld, O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J. Neuroimmunol. 2011, 232, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Dellacasa-Lindberg, I.; Hitziger, N.; Barragan, A. Localized recrudescence of Toxoplasma infections in the central nervous system of immunocompromised mice assessed by in vivo bioluminescence imaging. Microbes Infect. 2007, 9, 1291–1298. [Google Scholar] [CrossRef]
- Konradt, C.; Ueno, N.; Christian, D.A.; Delong, J.H.; Pritchard, G.H.; Herz, J.; Bzik, D.J.; Koshy, A.A.; McGavern, D.B.; Lodoen, M.B.; et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 2016, 1, 16001. [Google Scholar] [CrossRef] [PubMed]
- Marcos, A.C.; Siqueira, M.; Alvarez-Rosa, L.; Cascabulho, C.M.; Waghabi, M.C.; Barbosa, H.S.; Adesse, D.; Stipursky, J. Toxoplasma gondii infection impairs radial glia differentiation and its potential to modulate brain microvascular endothelial cell function in the cerebral cortex. Microvasc. Res. 2020, 131, 104024. [Google Scholar] [CrossRef] [PubMed]
- Franklin-Murray, A.L.; Mallya, S.; Jankeel, A.; Sureshchandra, S.; Messaoudi, I.; Lodoen, M.B. Toxoplasma gondii dysregulates barrier function and mechanotransduction signaling in human endothelial cells. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Kuraishy, H.M.; Al-Kuraishi, A.H.; Al-Windy, S.; Al-Gareeb, A.I. Toxoplasmosis and risk of endothelial dysfunction: Role of oxidative stress and pro-inflammatory mediators. Arch. Clin. Infect. Dis. 2019, 14, e95563. [Google Scholar] [CrossRef] [Green Version]
- Estato, V.; Stipursky, J.; Gomes, F.; Mergener, T.C.; Frazao-Teixeira, E.; Allodi, S.; Tibirica, E.; Barbosa, H.S.; Adesse, D. The neurotropic parasite Toxoplasma gondii induces sustained neuroinflammation with microvascular dysfunction in infected mice. Am. J. Pathol. 2018, 188, 2674–2687. [Google Scholar] [CrossRef] [Green Version]
- Portillo, J.C.; Van Grol, J.; Saffo, S.; Lopez Corcino, Y.; Rodriguez, M.; Fox, B.A.; Bzik, D.J.; Ward, N.L.; Dubyak, G.R.; Rojas, R.E.; et al. CD40 in endothelial cells restricts neural tissue invasion by Toxoplasma gondii. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Elsheikha, H.M.; McKinlay, C.L.; Elsaied, N.A.; Smith, P.A. Effects of Neospora caninum infection on brain microvascular endothelial cells bioenergetics. Parasit. Vectors 2013, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Elsheikha, H.M.; Alkurashi, M.; Kong, K.; Zhu, X.Q. Metabolic footprinting of extracellular metabolites of brain endothelium infected with Neospora caninum in vitro. BMC Res. Notes 2014, 7, 406. [Google Scholar] [CrossRef] [Green Version]
- Elsheikha, H.M.; Rosenthal, B.M.; Murphy, A.J.; Dunams, D.B.; Neelis, D.A.; Mansfield, L.S. Generally applicable methods to purify intracellular coccidia from cell cultures and to quantify purification efficacy using quantitative PCR. Vet. Parasitol. 2006, 135, 223–234. [Google Scholar] [CrossRef]
- Elsheikha, H.M.; Elsaied, N.A.; Chan, K.L.A.; Brignell, C.; Harun, M.S.R.; Wehbe, K.; Cinquee, G. Label-free characterization of biochemical changes within human cells under parasite attack using synchrotron based micro-FTIR. Anal. Methods 2019, 11, 2518–2530. [Google Scholar] [CrossRef]
- Shen, G.; Lesnoff, M.; Baeten, V.; Dardenne, P.; Davrieux, F.; Ceballos, H.; Belalcazar, J.; Dufour, D.; Yang, Z.; Han, L.; et al. Local partial least squares based on global PLS scores. J. Chemom. 2019, 33, e3117. [Google Scholar] [CrossRef]
- Charron, A.J.; Sibley, L.D. Host cells: Mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. J. Cell Sci. 2002, 115, 3049–3059. [Google Scholar] [PubMed]
- Naemat, A.; Elsheikha, H.M.; Al-Sandaqchi, A.; Kong, K.; Ghita, A.; Notingher, I. Analysis of interaction between the apicomplexan protozoan Toxoplasma gondii and host cells using label-free Raman spectroscopy. Analyst 2015, 140, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Naemat, A.; Elsheikha, H.M.; Boitor, R.A.; Notingher, I. Tracing amino acid exchange during host-pathogen interaction by combined stable-isotope time-resolved Raman spectral imaging. Sci. Rep. 2016, 6, 20811. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.X.; Zhou, D.H.; Elsheikha, H.M.; Liu, G.X.; Suo, X.; Zhu, X.Q. Global metabolomic profiling of mice brains following experimental infection with the cyst-forming Toxoplasma gondii. PLoS ONE 2015, 10, e0139635. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.X.; Zhou, D.H.; Elsheikha, H.M.; Zhao, Y.; Suo, X.; Zhu, X.Q. Metabolomic profiling of mice serum during toxoplasmosis progression using liquid chromatography-mass spectrometry. Sci. Rep. 2016, 6, 19557. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.D.; Catterall, J.R.; Remington, J.S. Parasiticidal activity of macrophages against Toxoplasma. Methods Enzymol. 1986, 132, 626–637. [Google Scholar] [CrossRef]
- Pelloux, H.; Pernod, G.; Polack, B.; Coursange, E.; Ricard, J.; Verna, J.M.; Ambroise-Thomas, P. Influence of cytokines on Toxoplasma gondii growth in human astrocytoma-derived cells. Parasitol. Res. 1996, 82, 598–603. [Google Scholar] [CrossRef]
- Barragan, A.; Brossier, F.; Sibley, L.D. Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell. Microbiol. 2005, 7, 561–568. [Google Scholar] [CrossRef]
- Furtado, J.M.; Bharadwaj, A.S.; Chipps, T.J.; Pan, Y.; Ashander, L.M.; Smith, J.R. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol. Cell. Biol. 2012, 90, 912–915. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.C.; Yeh, S.; Hirsch, J.G. The interaction between Toxoplasma gondii and mammalian cells. I. Mechanism of entry and intracellular fate of the parasite. J. Exp. Med. 1972, 136, 1157–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumdar, J.; Striepen, B. Make it or take it: Fatty acid metabolism of apicomplexan parasites. Eukaryot. Cell 2007, 6, 1727–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomavo, S.; Schwarz, R.T.; Dubremetz, J.F. Evidence for glycosyl-phosphatidylinositol anchoring of Toxoplasma gondii major surface antigens. Mol. Cell. Biol. 1989, 9, 4576–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, Y.; Quittnat, F.; Stedman, T.T.; Voelker, D.R.; Choi, J.Y.; Zahn, M.; Yang, M.; Pypaert, M.; Joiner, K.A.; Coppens, I. Host cell lipids control cholesteryl ester synthesis and storage in intracellular Toxoplasma. Cell. Microbiol. 2005, 7, 849–867. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Ibrahim, H.M.; Kameyama, K.; Shiga, I.; Hiasa, J.; Xuan, X. Host cholesterol synthesis contributes to growth of intracellular Toxoplasma gondii in macrophages. J. Vet. Med. Sci. 2011, 73, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffaro, C.E.; Boothroyd, J.C. Evidence for host cells as the major contributor of lipids in the intravacuolar network of Toxoplasma-infected cells. Eukaryot. Cell 2011, 10, 1095–1099. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.F.; Magalhaes, K.G.; Rodrigues, R.M.; de Carvalho, L.; Molinaro, R.; Bozza, P.T.; Barbosa, H.S. Toxoplasma gondii-skeletal muscle cells interaction increases lipid droplet biogenesis and positively modulates the production of IL-12, IFN-g and PGE2. Parasit. Vectors 2014, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, A.R.; Leve, F.; Morgado-Diaz, J.; Tedesco, R.C.; Pereira, M.C. Effect of Toxoplasma gondii infection on the junctional complex of retinal pigment epithelial cells. Parasitology 2016, 143, 568–575. [Google Scholar] [CrossRef]
- Briceno, M.P.; Nascimento, L.A.; Nogueira, N.P.; Barenco, P.V.; Ferro, E.A.; Rezende-Oliveira, K.; Goulart, L.R.; Alves, P.T.; Barbosa Bde, F.; Lima, W.R.; et al. Toxoplasma gondii Infection Promotes Epithelial Barrier Dysfunction of Caco-2 Cells. J. Histochem. Cytochem. 2016, 64, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Mayhan, W.G. Regulation of blood-brain barrier permeability. Microcirculation 2001, 8, 89–104. [Google Scholar] [CrossRef]
- Rao, R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 2008, 13, 7210–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, K.; Kakigawa, N.; Sekine, S.; Shitara, Y.; Horie, T. Disruption of ZO-1/claudin-4 interaction in relation to inflammatory responses in methotrexate-induced intestinal mucositis. Cancer Chemother. Pharmacol. 2013, 72, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Lavine, M.D.; Arrizabalaga, G. Exit from host cells by the pathogenic parasite Toxoplasma gondii does not require motility. Eukaryot. Cell 2008, 7, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blader, I.J.; Koshy, A.A. Toxoplasma gondii development of its replicative niche: In its host cell and beyond. Eukaryot. Cell 2014, 13, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzsche, R.; Zagoriy, V.; Lucius, R.; Gupta, N. Metabolic cooperation of glucose and glutamine is essential for the lytic cycle of obligate intracellular parasite Toxoplasma gondii. J. Biol. Chem. 2016, 291, 126–141. [Google Scholar] [CrossRef] [Green Version]
- Tielens, A.G.; van Grinsven, K.W.; Henze, K.; van Hellemond, J.J.; Martin, W. Acetate formation in the energy metabolism of parasitic helminths and protists. Int. J. Parasitol. 2010, 40, 387–397. [Google Scholar] [CrossRef]
- Syn, G.; Anderson, D.; Blackwell, J.M.; Jamieson, S.E. Toxoplasma gondii infection is associated with mitochondrial dysfunction in-vitro. Front. Cell. Infect. Microbiol. 2017, 7, 512. [Google Scholar] [CrossRef] [Green Version]
- Lang-Unnasch, N.; Murphy, A.D. Metabolic changes of the malaria parasite during the transition from the human to the mosquito host. Annu. Rev. Microbiol. 1998, 52, 561–590. [Google Scholar] [CrossRef]
- Mehta, M.; Sonawat, H.M.; Sharma, S. Malaria parasite-infected erythrocytes inhibit glucose utilization in uninfected red cells. FEBS Lett. 2005, 579, 6151–6158. [Google Scholar] [CrossRef] [Green Version]
- Lamour, S.D.; Straschil, U.; Saric, J.; Delves, M.J. Changes in metabolic phenotypes of Plasmodium falciparum in vitro cultures during gametocyte development. Malar. J. 2014, 13, 468. [Google Scholar] [CrossRef] [Green Version]
- Ohsaka, A.; Yoshikawa, K.; Hagiwara, T. 1H-NMR spectroscopic study of aerobic glucose metabolism in Toxoplasma gondii harvested from the peritoneal exudate of experimentally infected mice. Physiol. Chem. Phys. 1982, 14, 381–384. [Google Scholar] [PubMed]
- Kim, K.; Weiss, L.M. Toxoplasma gondii: The model apicomplexan. Int. J. Parasitol. 2004, 34, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Dubremetz, J.F.; Lebrun, M. Virulence factors of Toxoplasma gondii. Microbes Infect. 2012, 14, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Sibley, L.D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 2012, 10, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.V.; Holmes, E.; Saric, J.; Keiser, J.; Dirnhofer, S.; Utzinger, J.; Wang, Y. Metabolic profiling of a Schistosoma mansoni infection in mouse tissues using magic angle spinning-nuclear magnetic resonance spectroscopy. Int. J. Parasitol. 2009, 39, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Camarda, R.; Lagunoff, M. Vaccinia virus requires glutamine but not glucose for efficient replication. J. Virol. 2014, 88, 4366–4374. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Zahn, M.M.; Coppens, I.; Joiner, K.A.; Voelker, D.R. Selective disruption of phosphatidylcholine metabolism of the intracellular parasite Toxoplasma gondii arrests its growth. J. Biol. Chem. 2005, 280, 16345–16353. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, T.B. Strategies for acquiring the phospholipid metabolite inositol in pathogenic bacteria, fungi and protozoa: Making it and taking it. Microbiology 2009, 155, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.F.; Holmes, E.; Xue, J.; Xiao, S.H.; Singer, B.H.; Tang, H.R.; Utzinger, J.; Wang, Y.L. Metabolic alterations in the hamster co-infected with Schistosoma japonicum and Necator americanus. Int. J. Parasitol. 2010, 40, 695–703. [Google Scholar] [CrossRef]
- Govindaraju, V.; Young, K.; Maudsley, A.A. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000, 13, 129–153. [Google Scholar] [CrossRef]
- Block, W.; Traber, F.; Flacke, S.; Jessen, F.; Pohl, C.; Schild, H. In-vivo proton MR-spectroscopy of the human brain: Assessment of N-acetylaspartate (NAA) reduction as a marker for neurodegeneration. Amino Acids 2002, 23, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, R.; Baudrimont, M.; Lionnet, F.; Salord, J.M.; Duvivier, C.; Michon, C.; Wolff, M.; Marche, C. Skeletal muscle toxoplasmosis in patients with acquired immunodeficiency syndrome: A clinical and pathological study. Ann. Neurol. 1992, 32, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.M.; Pongponratan, E.; Supanaranond, W.; Pukrittayakamee, S.; Helliwell, T.; Holloway, P.; White, N.J. Skeletal muscle involvement in falciparum malaria: Biochemical and ultrastructural study. Clin. Infect. Dis. 1999, 29, 831–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harun, M.S.R.; Marsh, V.; Elsaied, N.A.; Webb, K.F.; Elsheikha, H.M. Effects of Toxoplasma gondii infection on the function and integrity of human cerebrovascular endothelial cells and the influence of verapamil treatment in vitro. Brain Res. 2020, 1746. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-sandaqchi, A.T.; Marsh, V.; Williams, H.E.L.; Stevenson, C.W.; Elsheikha, H.M. Structural, Functional, and Metabolic Alterations in Human Cerebrovascular Endothelial Cells during Toxoplasma gondii Infection and Amelioration by Verapamil In Vitro. Microorganisms 2020, 8, 1386. https://doi.org/10.3390/microorganisms8091386
Al-sandaqchi AT, Marsh V, Williams HEL, Stevenson CW, Elsheikha HM. Structural, Functional, and Metabolic Alterations in Human Cerebrovascular Endothelial Cells during Toxoplasma gondii Infection and Amelioration by Verapamil In Vitro. Microorganisms. 2020; 8(9):1386. https://doi.org/10.3390/microorganisms8091386
Chicago/Turabian StyleAl-sandaqchi, Alaa T., Victoria Marsh, Huw E. L. Williams, Carl W. Stevenson, and Hany M. Elsheikha. 2020. "Structural, Functional, and Metabolic Alterations in Human Cerebrovascular Endothelial Cells during Toxoplasma gondii Infection and Amelioration by Verapamil In Vitro" Microorganisms 8, no. 9: 1386. https://doi.org/10.3390/microorganisms8091386