Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities Using Different Primer Sets on the Loess Plateau


Abstract
:1. Introduction
2. Methods and Materials
2.1. Site Description and Sampling
2.2. Climate and Edaphic Characteristics
2.3. DNA Extraction
2.4. 16S rRNA Gene Amplification and Sequencing
2.5. Processing of the Sequencing Data
2.6. Statistical Analysis
3. Results
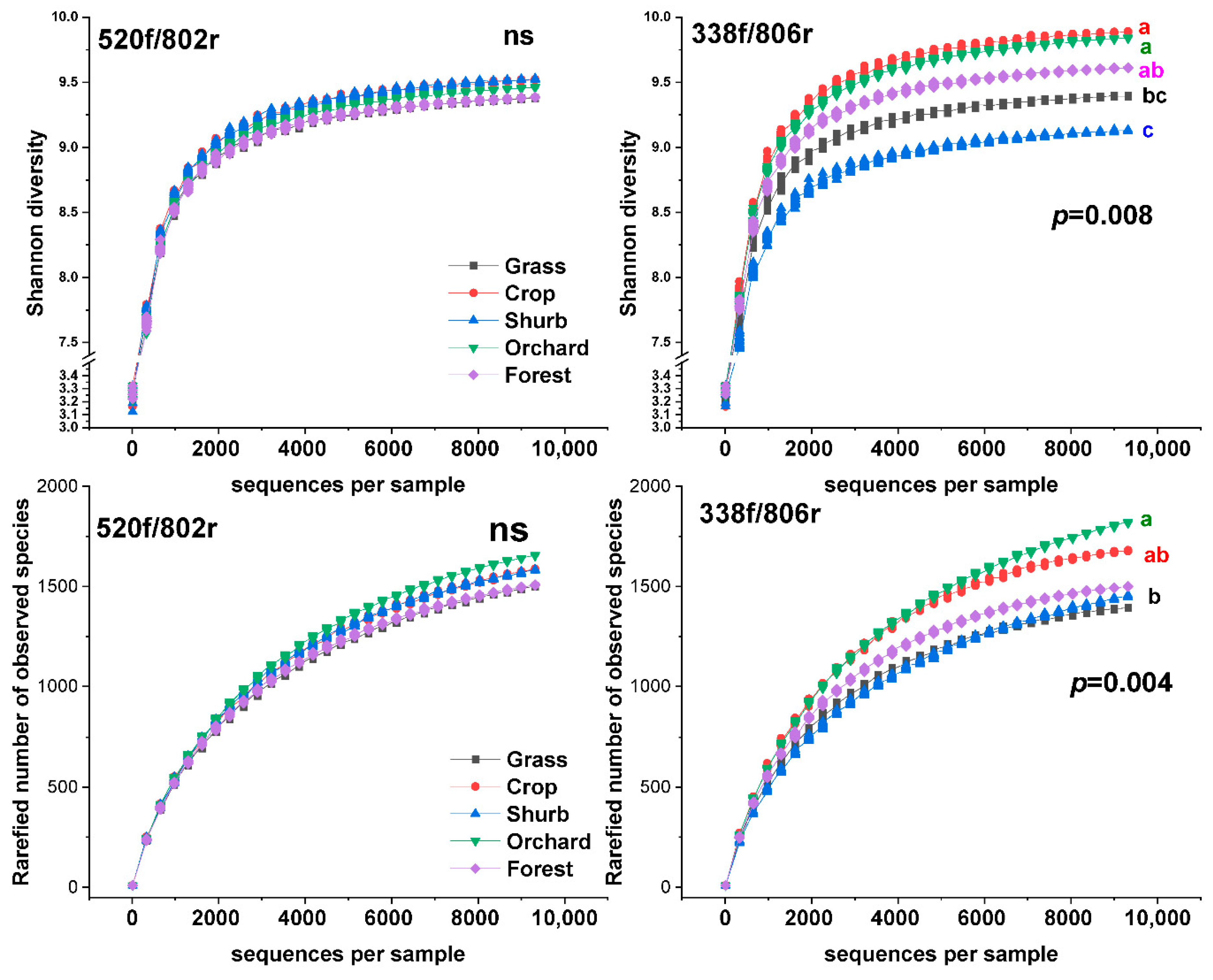
3.1. The Distributions of the Soil Bacterial Diversity
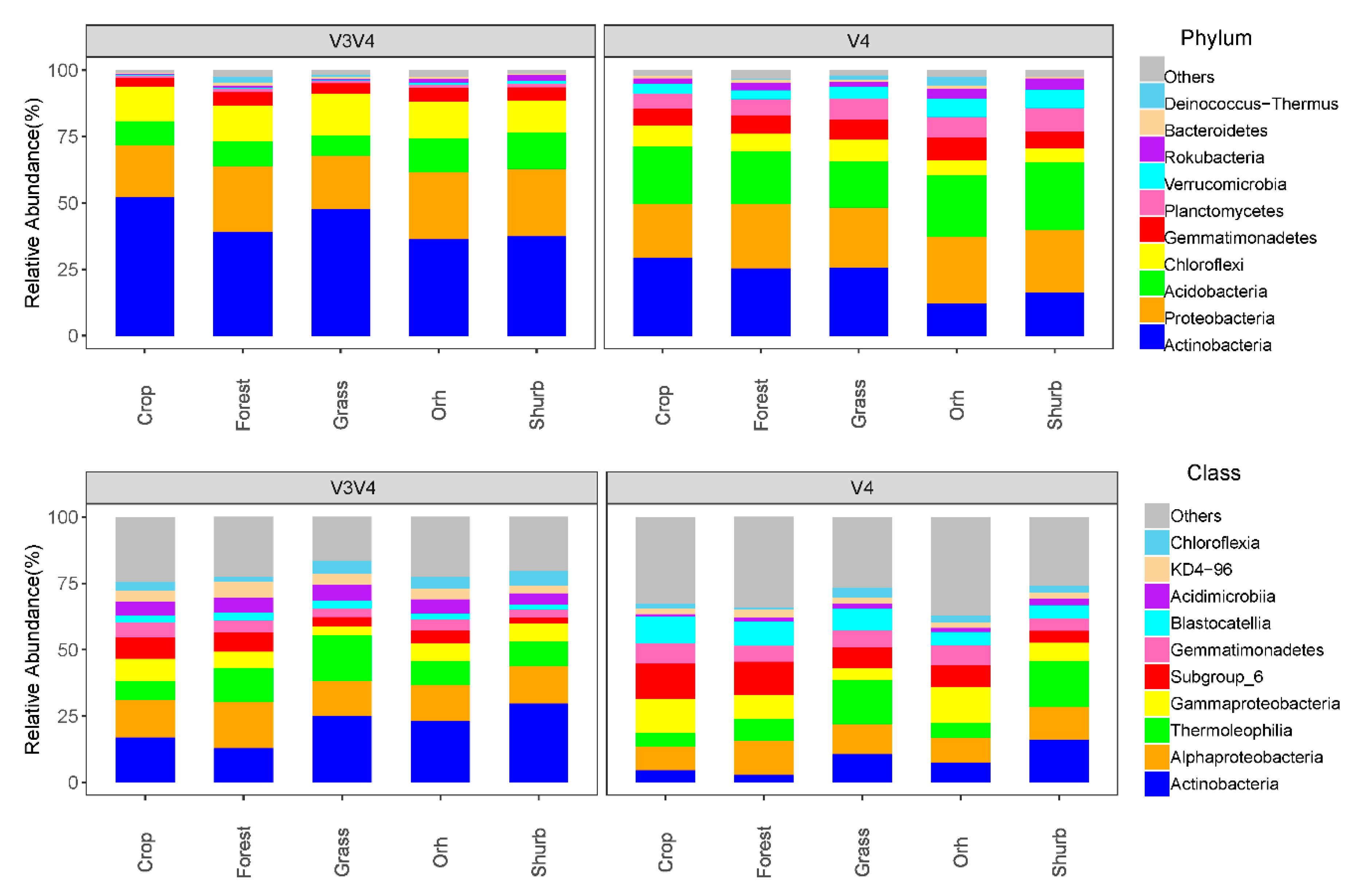
3.2. The Distributions of the Soil Bacterial Community Compositions
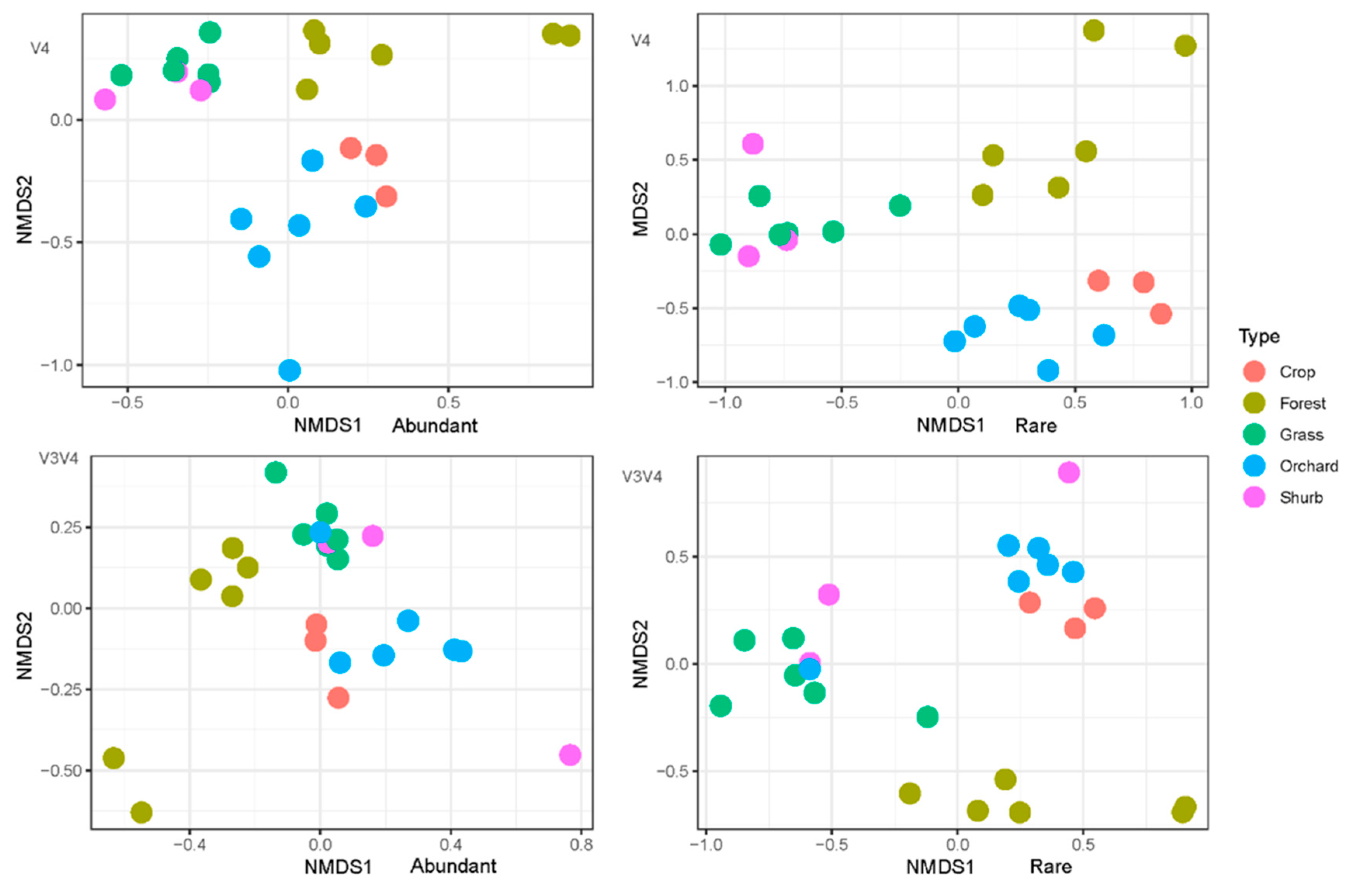
3.3. The Variations in the Soil Bacterial Community Structure
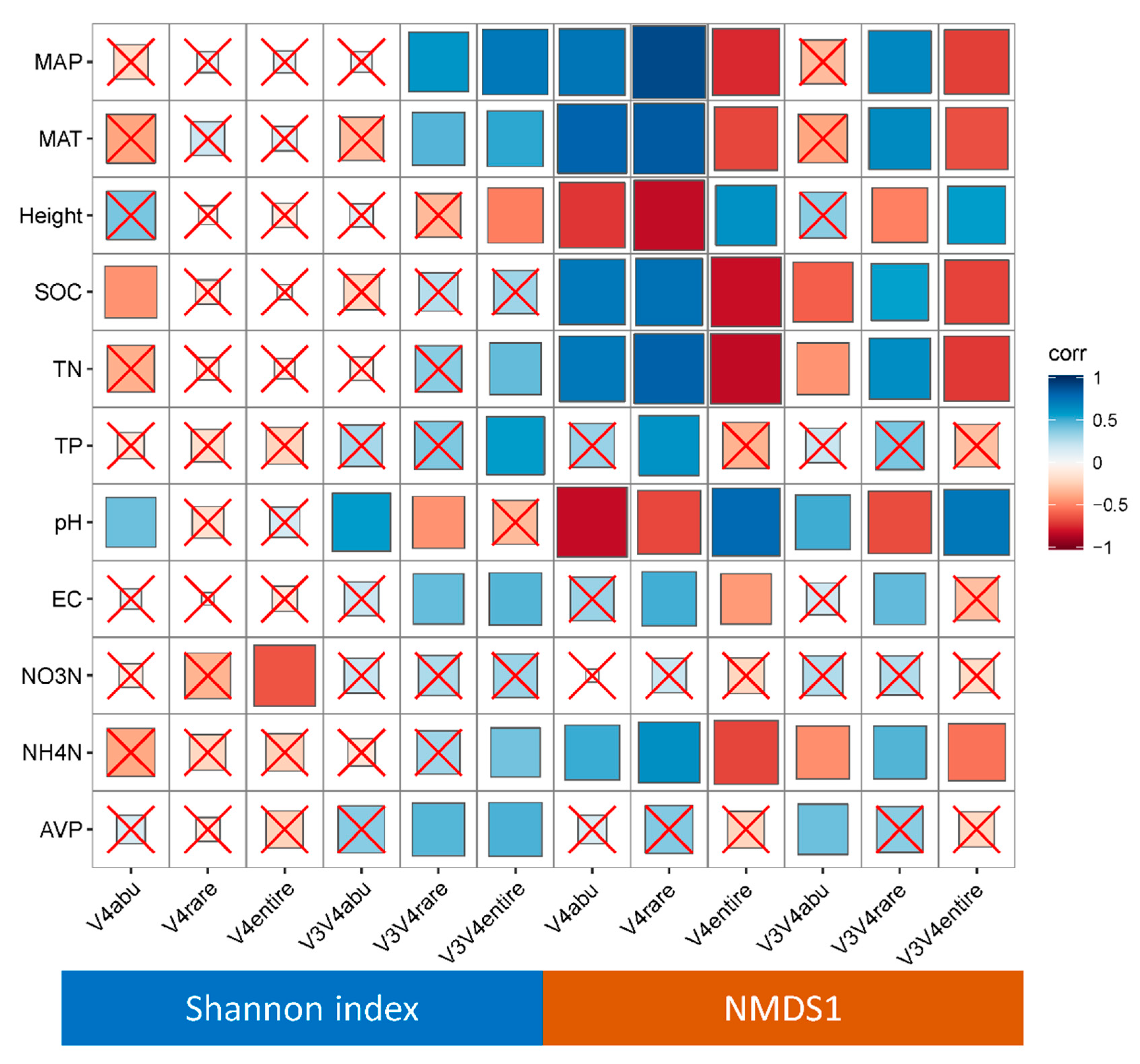
3.4. Geographic Distance and Environmental Factors Regulate Bacterial Community Structure
4. Discussion
4.1. Land Use Affected the Soil Microbial Diversity
4.2. Land Use Affected the Soil Microbial Community Compositions
4.3. The Role of Environmental Factors in Shaping Soil Bacterial Community Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kurm, V.; van der Putten, W.H.; de Boer, W.; Naus-Wiezer, S.; Hol, W.H.G. Low abundant soil bacteria can be metabolically versatile and fast growing. Ecology 2017, 98, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausmann, B.; Knorr, K.-H.; Schreck, K.; Tringe, S.G.; Glavina del Rio, T.; Loy, A.; Pester, M. Consortia of low-abundance bacteria drive sulfate reduction-dependent degradation of fermentation products in peat soil microcosms. ISME J. 2016, 10, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-L.; Ding, J.; Zhu, D.; Hu, H.-W.; Delgado-Baquerizo, M.; Ma, Y.-B.; He, J.-Z.; Zhu, Y.-G. Rare microbial taxa as the major drivers of ecosystem multifunctionality in long-term fertilized soils. Soil Biol. Biochem. 2020, 141, 107686. [Google Scholar] [CrossRef]
- Li, P.; Liu, J.; Jiang, C.; Wu, M.; Liu, M.; Li, Z. Distinct successions of common and rare bacteria in soil under humic acid amendment—A microcosm study. Front. Microbiol. 2019, 10, 2271. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Chen, W.; Wei, G. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 2017, 26, 5305–5317. [Google Scholar] [CrossRef]
- Liang, Y.; Xiao, X.; Nuccio, E.E.; Yuan, M.; Zhang, N.; Xue, K.; Cohan, F.M.; Zhou, J.; Sun, B. Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environ. Microbiol. 2020, 22, 1327–1340. [Google Scholar] [CrossRef]
- Jiao, S.; Lu, Y. Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems. Environ. Microbiol. 2020, 22, 1052–1065. [Google Scholar] [CrossRef]
- Ji, M.; Kong, W.; Stegen, J.; Yue, L.; Wang, F.; Dong, X.; Don Cowan, A.; Ferrari, B.C. Distinct assembly mechanisms underlie similar biogeographical patterns of rare and abundant bacteria in Tibetan Plateau grassland soils. Environ. Microbiol. 2020, 22, 2261–2272. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Li, M.; Xu, L.; He, N.; Yan, P.; Chen, C.; Lu, Q.; Feng, Y.; Liet, J. Differential response of abundant and rare bacterial subcommunities to abiotic and biotic gradients across temperate deserts. Sci. Total Environ. 2020, 142942. [Google Scholar] [CrossRef]
- Logares, R.; Audic, S.; Bass, D.; Bittner, L.; Boutte, C.; Christen, R.; Claverie, J.-M.; Decelle, J.; Dolan, J.R.; Dunthorn, M.; et al. Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol. 2014, 24, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-S.; Jeong, J.-Y.; Wells, G.F.; Park, H.-D. General and rare bacterial taxa demonstrating different temporal dynamic patterns in an activated sludge bioreactor. Appl. Microbiol. Biotechnol. 2013, 97, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [Green Version]
- Ghyselinck, J.; Pfeiffer, S.; Heylen, K.; Sessitsch, A.; De Vos, P. The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS ONE 2013, 8, e71360. [Google Scholar]
- Soergel, D.A.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008, 36, e120. [Google Scholar] [CrossRef] [Green Version]
- Youssef, N.; Sheik, C.S.; Krumholz, L.R.; Najar, F.Z.; Roe, B.A.; Elshahed, M.S. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 2009, 75, 5227–5236. [Google Scholar] [CrossRef] [Green Version]
- Thijs, S.; Op De Beeck, M.; Beckers, B.; Truyens, S.; Stevens, V.; Van Hamme, J.D.; Weyens, N.; Vangronsveld, J. Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Front. Microbiol. 2017, 8, 494. [Google Scholar] [CrossRef]
- Vitousek, P.M.; Aber, J.D.; Howarth, R.W.; Likens, G.E.; Matson, P.A.; Schindler, D.W.; Schlesinger, W.H.; Tilman, D.G. Human alteration of the global nitrogen cycle: Sources and consequences. Ecol. Appl. 1997, 7, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Delgado-Baquerizo, M.; Guo, X.; Wang, D.; Wu, Y.; Zhu, M.; Yu, W.; Yao, H.; Zhu, Y.-G.; Chu, H. Suppressed N fixation and diazotrophs after four decades of fertilization. Microbiome 2019, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhang, M. Effects of land-use conversion from paddy field to orchard farm on soil microbial genetic diversity and community structure. Eur. J. Soil Biol. 2014, 64, 30–39. [Google Scholar] [CrossRef]
- Li, F.-R.; Feng, Q.; Liu, J.-L.; Sun, T.-S.; Ren, W.; Guan, Z.-H. Effects of the conversion of native vegetation to farmlands on soil microarthropod biodiversity and ecosystem functioning in a desert oasis. Ecosystems 2013, 16, 1364–1377. [Google Scholar] [CrossRef]
- Schwinning, S.; Sala, O.E. Hierarchy of responses to resource pulses in arid and semi-arid ecosystems. Oecologia 2004, 141, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Trivedi, P.; Osanai, Y.; Liu, Y.-R.; Hamonts, K.; Jeffries, T.C.; Singh, B.K. Carbon content and climate variability drive global soil bacterial diversity patterns. Ecol. Monogr. 2016, 86, 373–390. [Google Scholar] [CrossRef]
- Liu, D.; Yang, Y.; An, S.; Wang, H.; Wang, Y. The biogeographical distribution of soil bacterial communities in the loess plateau as revealed by high-throughput sequencing. Front. Microbiol. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Li, X.; Dong, Y.; An, S.; Darboux, F. Soil and plant components ecological stoichiometry in four steppe communities in the Loess Plateau of China. Catena 2016, 147, 481–488. [Google Scholar] [CrossRef]
- Zeng, Q.C.; An, S.S.; Liu, Y. Soil bacterial community response to vegetation succession after fencing in the grassland of China. Sci. Total Environ. 2017, 609, 2–10. [Google Scholar] [CrossRef]
- Ren, H.; Xu, Z.; Huang, J.; Lü, X.; Zeng, D.-H.; Yuan, Z.; Fang, Y.; Han, X.-G. Increased precipitation induces a positive plant-soil feedback in a semi-arid grassland. Plant Soil 2015, 389, 211–223. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berglyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkilä, J.; Marchesi, J.R.; Smidt, H.; de Vos Willem, M.; Ross, R.P.; O’Toole, P.W. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 2009, 4, e6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymoset, P.; Stevens, M.H.H.; Wagner, H.H. Package ‘vegan’. Community Ecol. Package Vers. 2013, 2, 1–295. [Google Scholar]
- Ripley, B.; Venables, B.; Bates, D.M.; Hornik, K.; Gebhardt, A.; Firth, D. Package ‘mass’. Cran R 2013. Available online: http://cran.r-projectorg/web/packages/MASS/MASS.pdf (accessed on 26 November 2020).
- Grömping, U. Estimators of relative importance in linear regression based on variance decomposition. Am. Statist. 2007, 61, 139–147. [Google Scholar] [CrossRef]
- Reinhart, K.O.; Dangi, S.R.; Vermeire, L.T. The effect of fire intensity, nutrients, soil microbes, and spatial distance on grassland productivity. Plant Soil 2016, 409, 203–216. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Chen, L.-F.; He, Z.-B.; Zhao, W.-Z.; Liu, J.-L.; Zhou, H.; Li, J.; Menget, Y.-Y.; Wang, L.-S. Soil structure and nutrient supply drive changes in soil microbial communities during conversion of virgin desert soil to irrigated cropland. Eur. J. Soil Sci. 2020, 71, 768–781. [Google Scholar] [CrossRef]
- Tian, Q.; Taniguchi, T.; Shi, W.-Y.; Li, G.; Yamanaka, N.; Du, S. Land-use types and soil chemical properties influence soil microbial communities in the semiarid Loess Plateau region in China. Sci. Rep. 2017, 7, 45289. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Fang, L.; Guo, X.; Wang, X.; Wang, Y.; Zhang, Y.; Zhang, C. Responses of soil bacterial communities, enzyme activities, and nutrients to agricultural-to-natural ecosystem conversion in the Loess Plateau, China. J. Soils Sedim. 2019, 19, 1427–1440. [Google Scholar] [CrossRef]
- Maestre, F.T.; Delgado-Baquerizo, M.; Jeffries, T.C.; Eldridge, D.J.; Ochoa, V.; Gozalo, B.; Quero, J.L.; García-Gómez, M.; Gallardo, A.; Ulrich, W.; et al. Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. USA 2015, 112, 15684–15689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.; McIntyre, N.; Cox, S.; Tissue, D.; Zak, J. Soil microbial responses to temporal variations of moisture and temperature in a chihuahuan desert grassland. Microb. Ecol. 2008, 56, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Maestre, F.T.; Gallardo, A.; Bowker, M.A.; Wallenstein, M.D.; Quero, J.L.; Ochoa, V.; Gozalo, B.; García-Gómez, M.; Soliveres, S.; et al. Decoupling of soil nutrient cycles as a function of aridity in global drylands. Nature 2013, 502, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Dong, Y.; An, S. Bacterial community responses to soils along a latitudinal and vegetation gradient on the Loess Plateau, China. PLoS ONE 2016, 11, e0152894. [Google Scholar]
- Yao, F.; Yang, S.; Wang, Z.; Wang, X.; Ye, J.; Wang, X.; DeBruyn, J.M.; Feng, X.; Jiang, Y.; Li, H. Microbial taxa distribution is associated with ecological trophic cascades along an elevation gradient. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- van der Bom, F.; Nunes, I.; Raymond, N.S.; Hansen, V.; Bonnichsen, L.; Magid, J.; Nybroe, O.; Jensen, L.S. Long-term fertilisation form, level and duration affect the diversity, structure and functioning of soil microbial communities in the field. Soil Biol. Biochem. 2018, 122, 91–103. [Google Scholar] [CrossRef]
- Rughöft, S.; Herrmann, M.; Lazar, C.S.; Cesarz, S.; Levick, S.R.; Trumbore, S.E.; Küsel, K. Community composition and abundance of bacterial, archaeal and nitrifying populations in savanna soils on contrasting bedrock material in Kruger National Park, South Africa. Front. Microbiol. 2016, 7, 1638. [Google Scholar]
- Tian, J.; He, N.; Hale, L.; Niu, S.; Yu, G.; Liu, Y.; Blagodatskaya, E.; Kuzyakov, Y.; Gao, Q.; Zhouet, J. Soil organic matter availability and climate drive latitudinal patterns in bacterial diversity from tropical to cold-temperate forests. Funct. Ecol. 2018, 32, 61–70. [Google Scholar] [CrossRef]
- Liu, J.; Sui, Y.; Yu, Z.; Shi, Y.; Chu, H.; Jin, J.; Liu, X.; Wanget, G. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China. Soil Biol. Biochem. 2014, 70, 113–122. [Google Scholar] [CrossRef]
- Zeng, Q.; An, S.; Liu, Y.; Wang, H.; Wang, Y. Biogeography and the driving factors affecting forest soil bacteria in an arid area. Sci. Total Environ. 2019, 680, 124–131. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, H.; Wang, R.; Guo, S.; Gao, C. Impact of root diversity upon coupling between soil C and N accumulation and bacterial community dynamics and activity: Result of a 30year rotation experiment. Geoderma 2017, 292, 87–95. [Google Scholar] [CrossRef]
- Trivedi, P.; Rochester, I.J.; Trivedi, C.; Van Nostrand, J.D.; Zhou, J.; Karunaratne, S.; Anderson, I.C.; Singh, B.K. Soil aggregate size mediates the impacts of cropping regimes on soil carbon and microbial communities. Soil Biol. Biochem. 2015, 91, 169–181. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wu, X.; Tai, X.; Sun, L.; Wu, M.; Zhang, W.; Chen, X.; Zhang, G.; Chen, T.; Liu, G.; et al. Variation in actinobacterial community composition and potential function in different soil ecosystems belonging to the arid Heihe River Basin of northwest China. Front. Microbiol. 2019, 10, 2209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.Z.; Fan, X.D.; Ren, C.J.; Zhang, L.; Han, X.H.; Yang, G.H.; Wang, J.; Doughty, R. Changes of the organic carbon content and stability of soil aggregates affected by soil bacterial community after afforestation. Catena 2018, 171, 622–631. [Google Scholar] [CrossRef]
- Wang, X.; Van Nostrand, J.D.; Deng, Y.; Lü, X.; Wang, C.; Zhou, J.; Han, X. Scale-dependent effects of climate and geographic distance on bacterial diversity patterns across northern China’s grasslands. FEMS Microbiol. Ecol. 2015, 91, fiv133. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Chu, H.; Fierer, N.; Lauber, C.L.; Caporaso, J.G.; Knight, R.; Grogan, P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. Rep. 2010, 12, 2998–3006. [Google Scholar] [CrossRef]
- Xia, Z.; Bai, E.; Wang, Q.; Gao, D.; Zhou, J.; Jiang, P.; Wu, J. Biogeographic distribution patterns of bacteria in typical Chinese forest soils. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, R.I.; Thomson, B.C.; James, P.; Bell, T.; Bailey, M.; Whiteley, A.S. The bacterial biogeography of British soils. Environ. Microbiol. 2011, 13, 1642–1654. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V3–V4 (Shannon Diversity) | Estimate | t | p | RI |
|---|---|---|---|---|
| (Intercept) | 6.727 | 14.906 | <0.0001 | |
| MAP | 0.005 | 5.803 | <0.0001 | 0.7029 |
| NH4N | 0.111 | 2.431 | 0.0246 | 0.1461 |
| SOC | −0.063 | −3.344 | 0.0032 | 0.1510 |
| V3–V4 (NMDS1) | Estimate | t | p | RI |
|---|---|---|---|---|
| (Intercept) | −0.645 | −1.373 | 0.186 | |
| pH | 0.171 | 3.4 | 0.003 | 0.4561 |
| SOC | −0.022 | −3.208 | 0.005 | 0.4103 |
| EC | 0.000 | 2.245 | 0.037 | 0.0742 |
| AVP | −0.005 | −2.333 | 0.031 | 0.0594 |
| V4 (NMDS1) | Estimate | t | p | RI |
|---|---|---|---|---|
| (Intercept) | −1.632 | −4.431 | 0.00023 | |
| pH | 0.143 | 3.598 | 0.00169 | 0.4598 |
| SOC | −0.025 | −4.432 | 0.00023 | 0.5402 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Q.; An, S. Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities Using Different Primer Sets on the Loess Plateau. Microorganisms 2021, 9, 139. https://doi.org/10.3390/microorganisms9010139
Zeng Q, An S. Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities Using Different Primer Sets on the Loess Plateau. Microorganisms. 2021; 9(1):139. https://doi.org/10.3390/microorganisms9010139
Chicago/Turabian StyleZeng, Quanchao, and Shaoshan An. 2021. "Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities Using Different Primer Sets on the Loess Plateau" Microorganisms 9, no. 1: 139. https://doi.org/10.3390/microorganisms9010139
APA StyleZeng, Q., & An, S. (2021). Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities Using Different Primer Sets on the Loess Plateau. Microorganisms, 9(1), 139. https://doi.org/10.3390/microorganisms9010139