Chlamydia Uses K+ Electrical Signalling to Orchestrate Host Sensing, Inter-Bacterial Communication and Differentiation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Procedures
2.1. Cell Culture and Infection
2.2. Drug Treatments
2.3. Persistence Induction
2.4. Flame Photometry
2.5. Fluorescence Labelling
2.6. Confocal Microscopy
2.7. Image Analysis
2.8. Infectivity Assay
2.9. rRNA Isolation and Amplification
2.10. Transmission Electron Microscopy (TEM)
3. Results
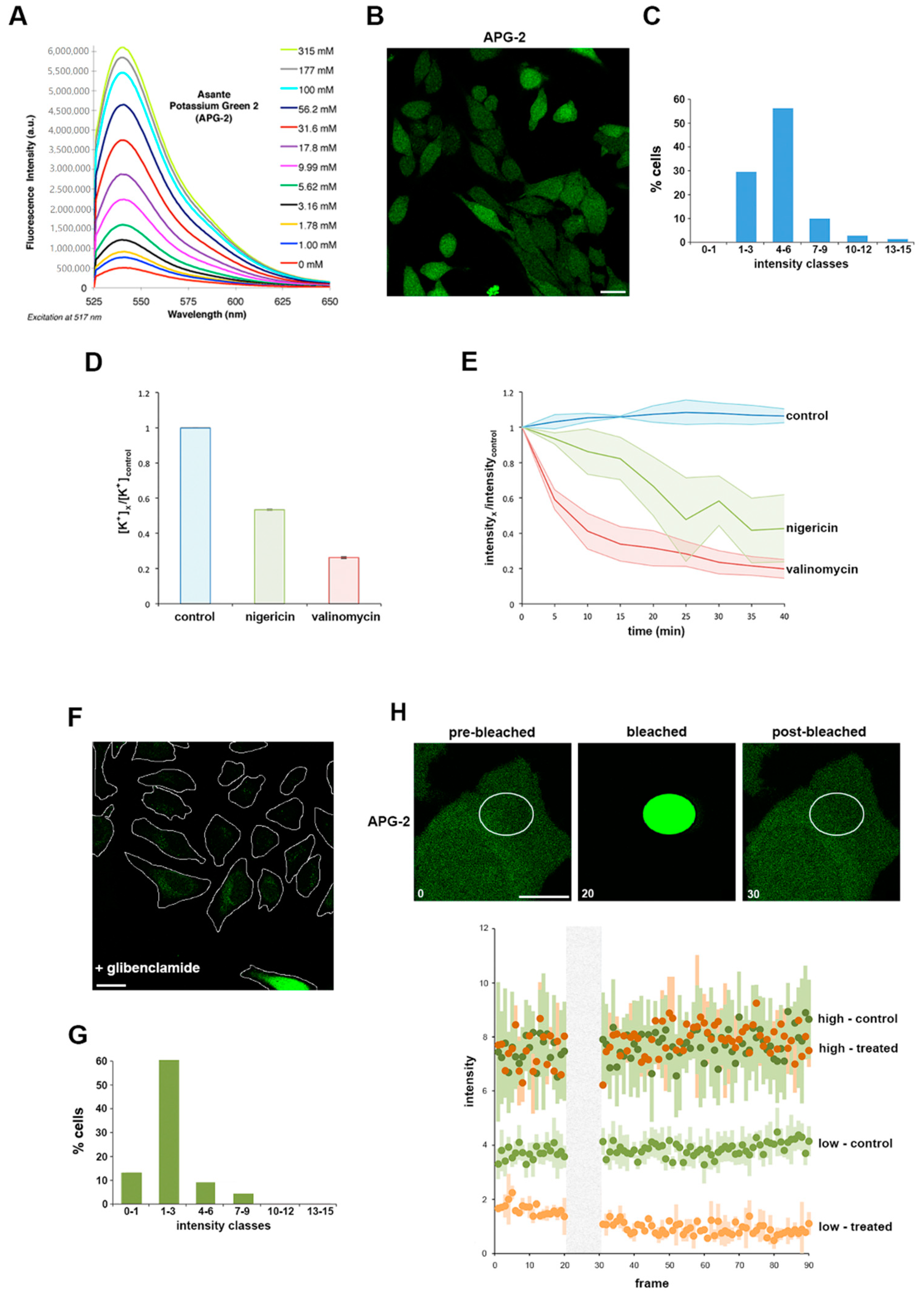
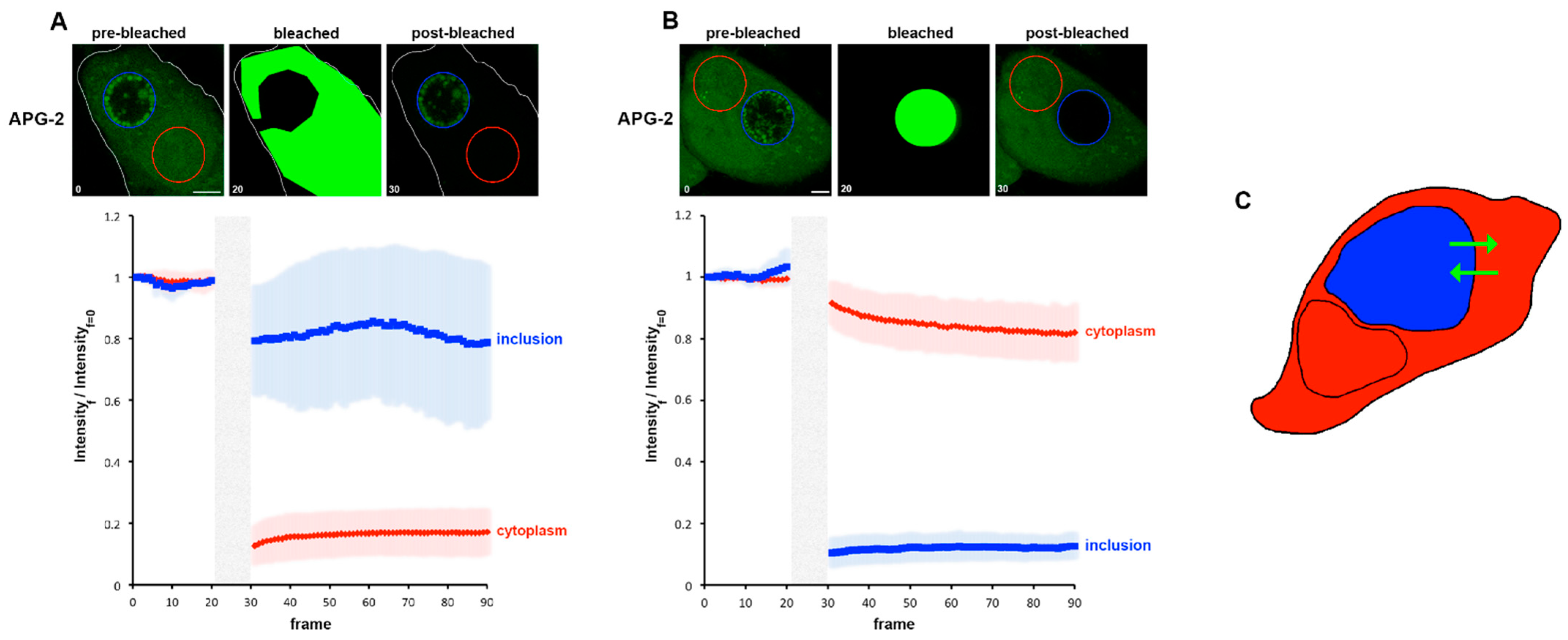
3.1. Using APG-2 as a Potassium Ion (K +) Probe for Live Cell Imaging
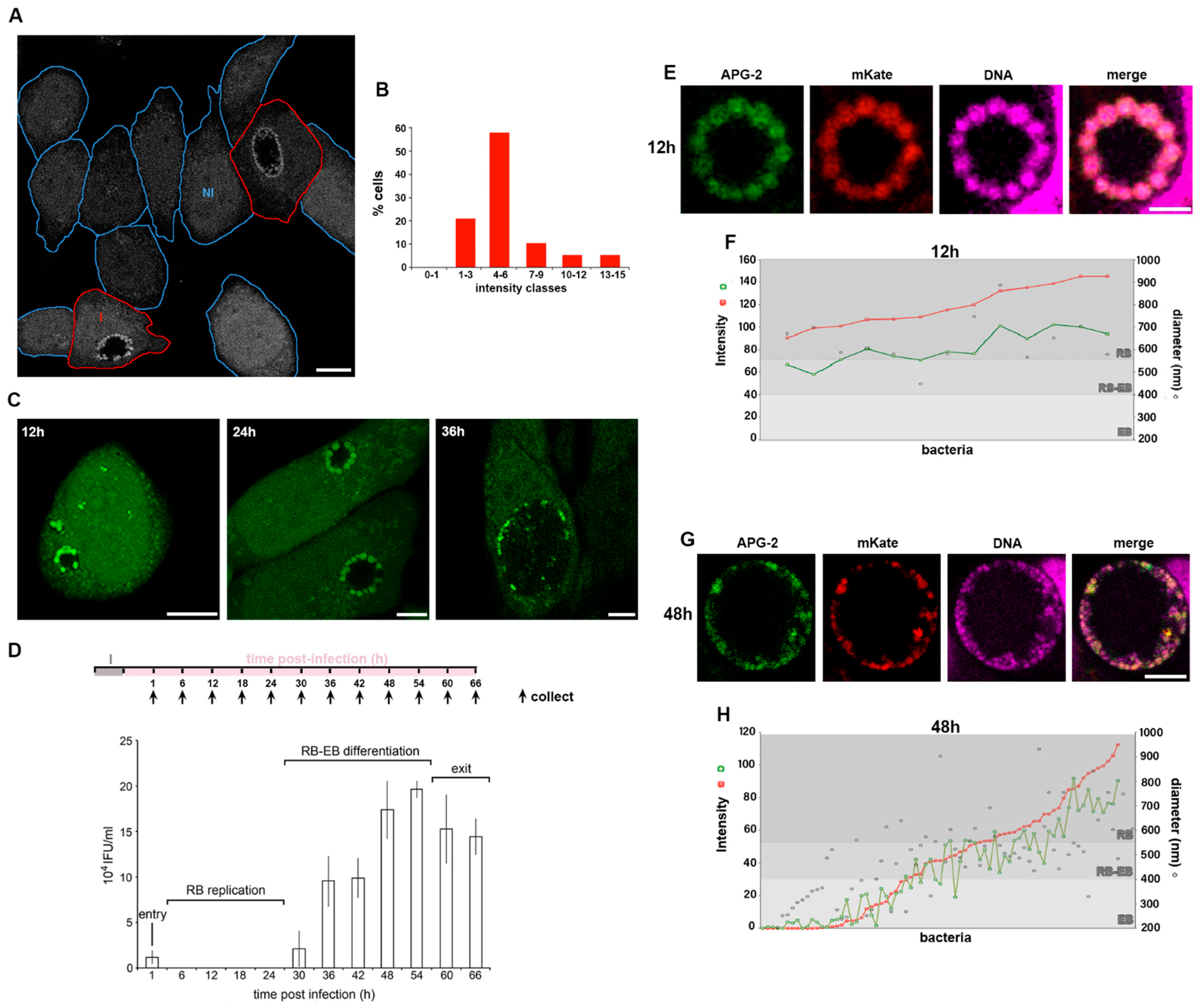
3.2. Potassium Ions Accumulate within Replicative C. trachomatis Reticulate Bodies
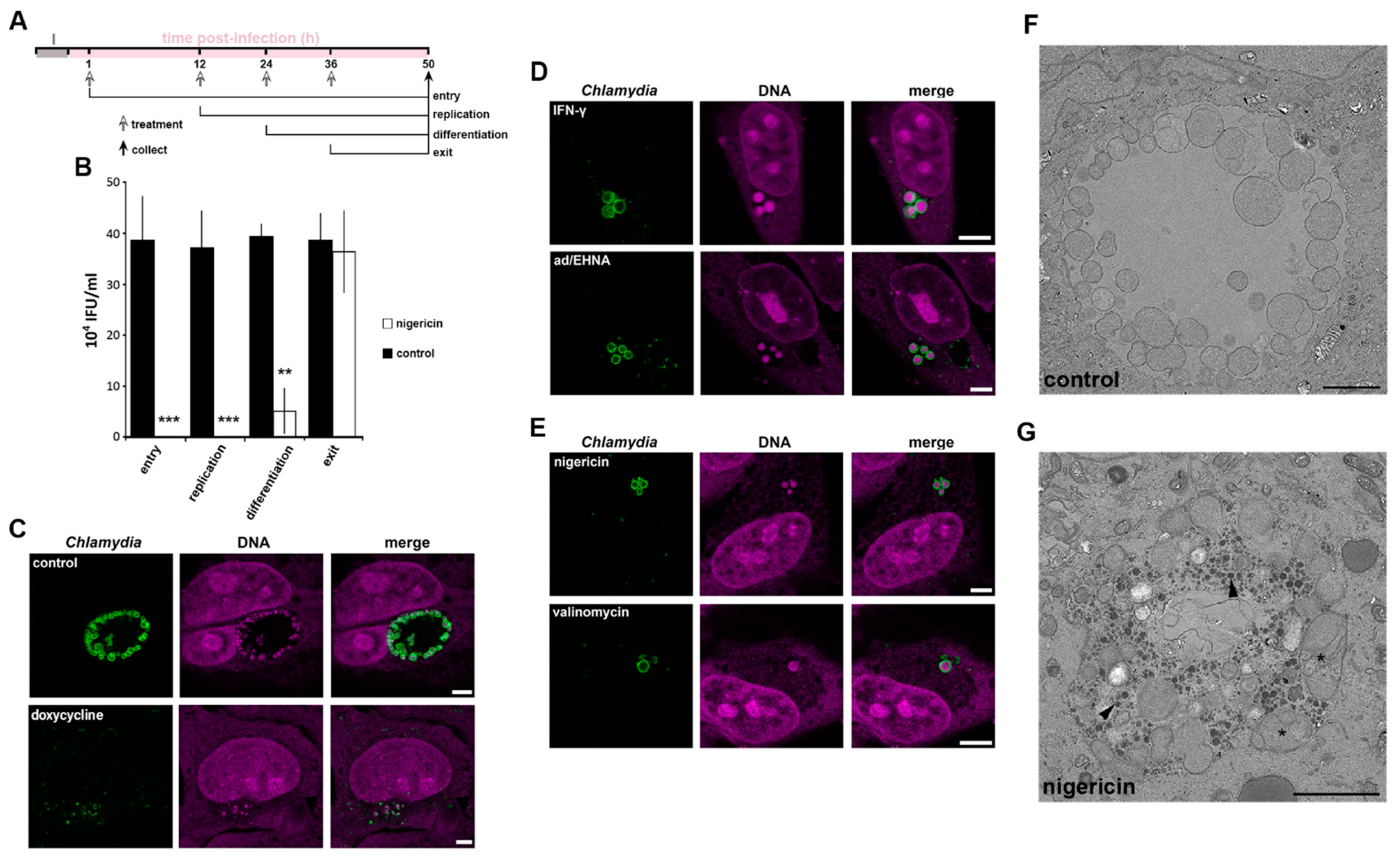
3.3. The Role of K+ in the Bacterial Stress Response
3.4. Disruption of [K+] Is a Common Aspect of Chlamydia Persistence
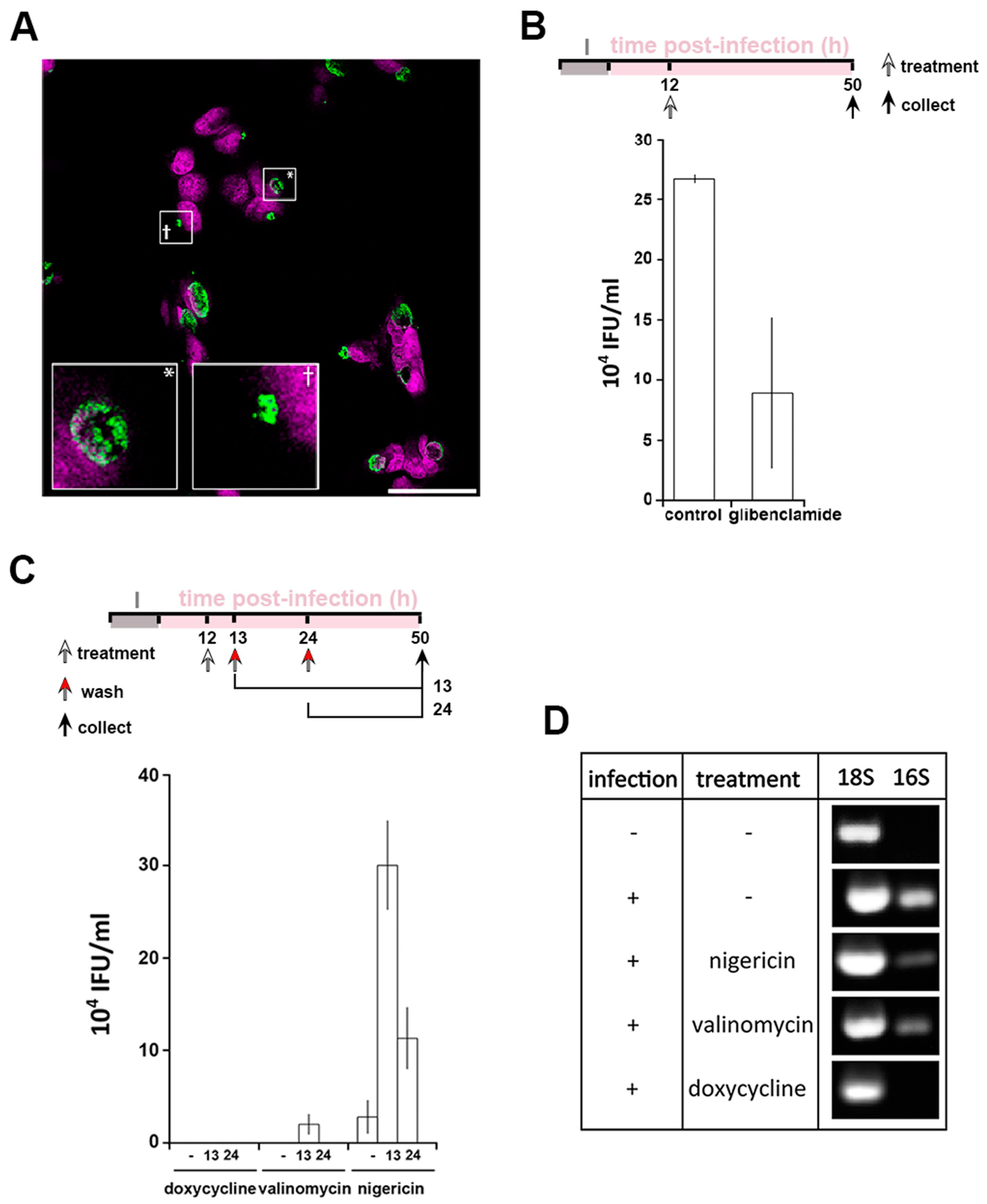
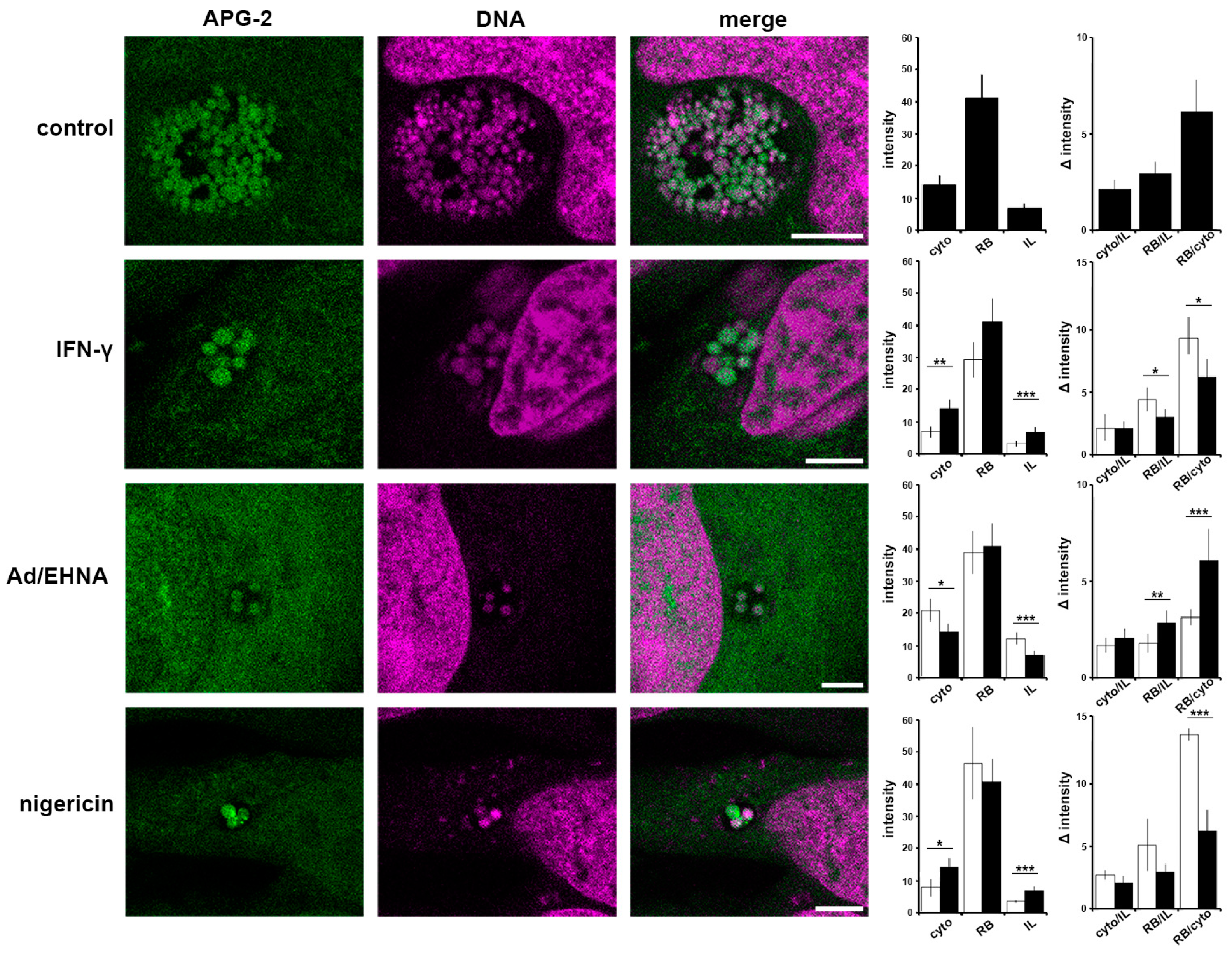
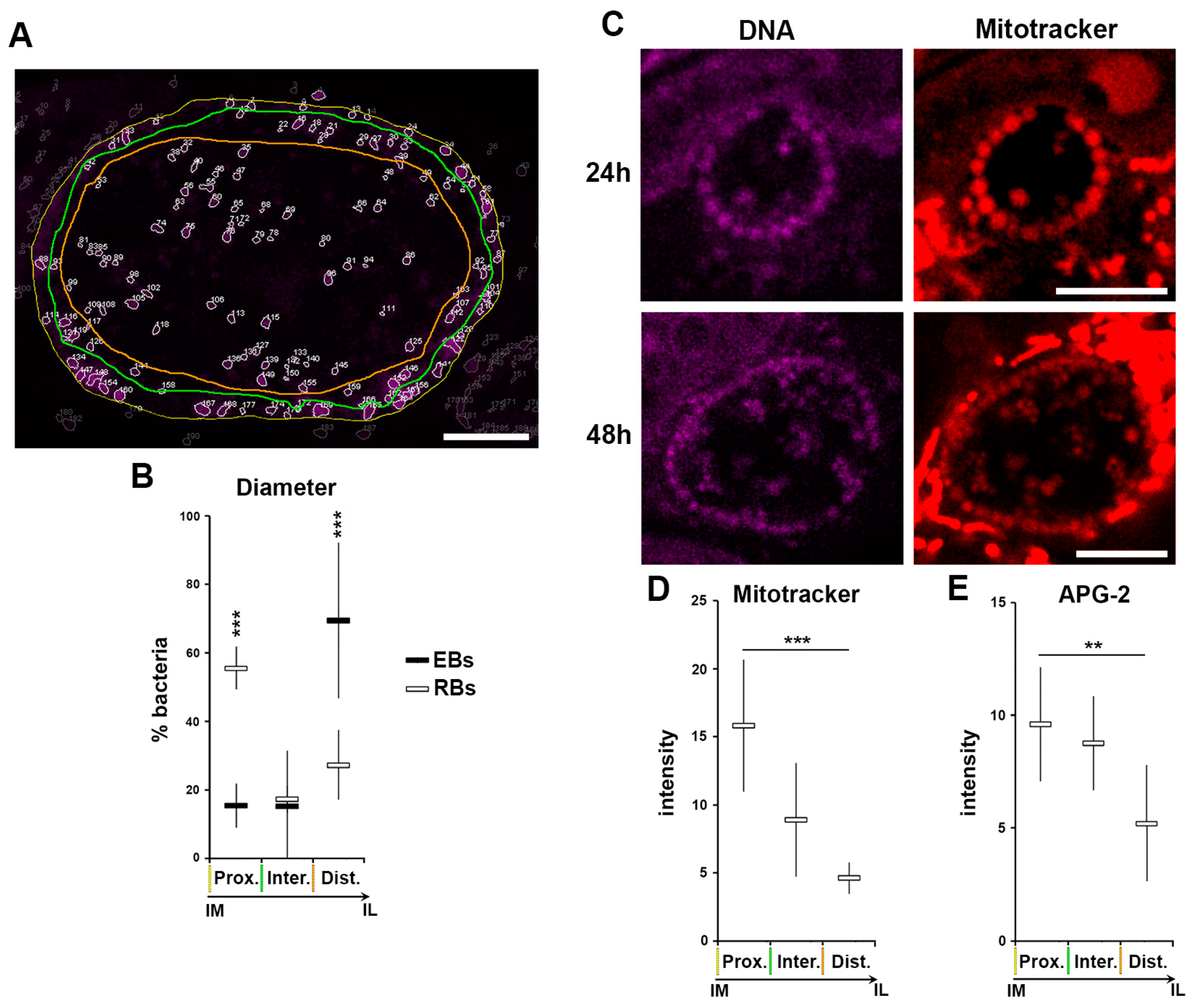
3.5. A Role for K+ in Host Pathogen Communication
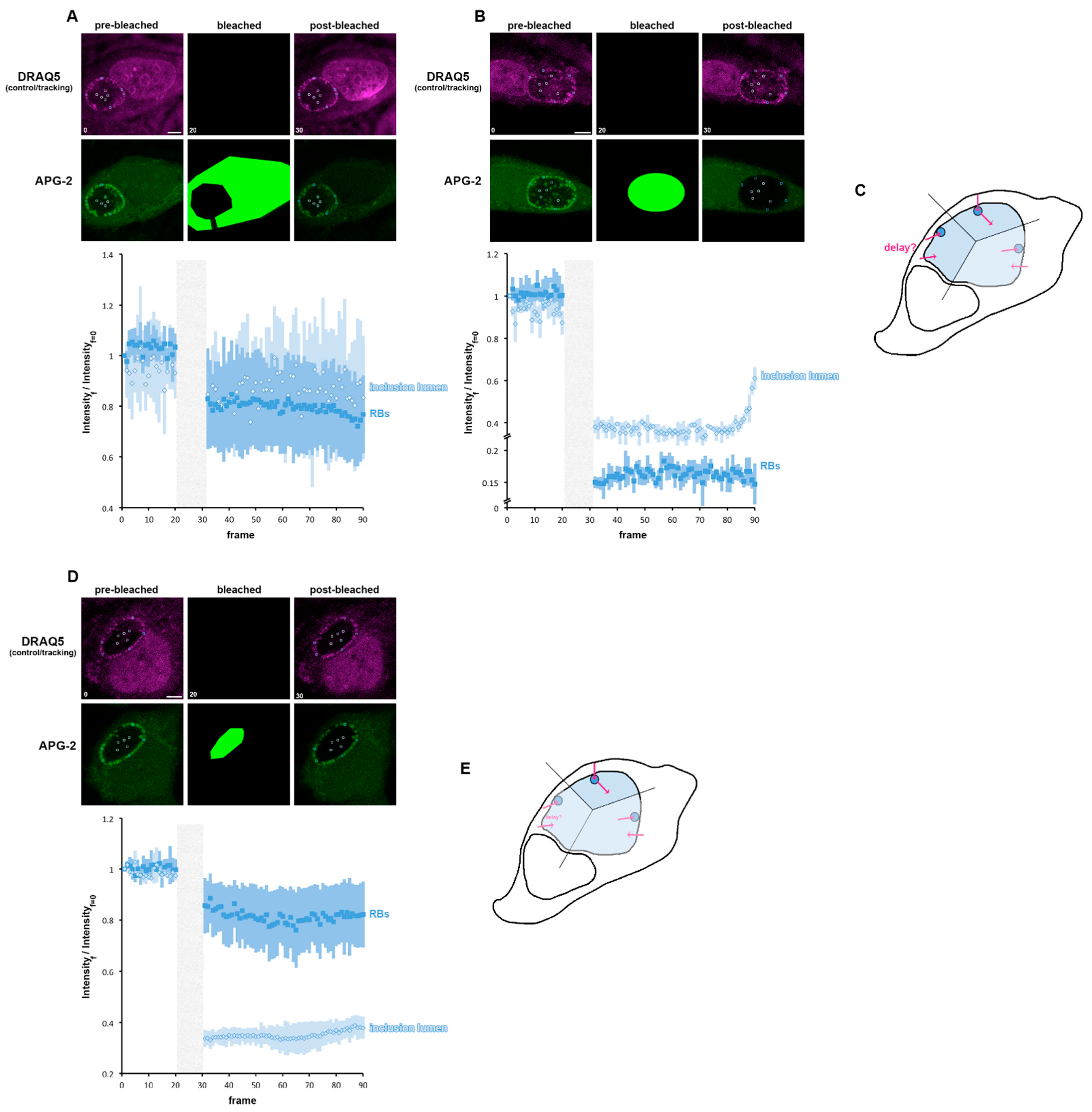
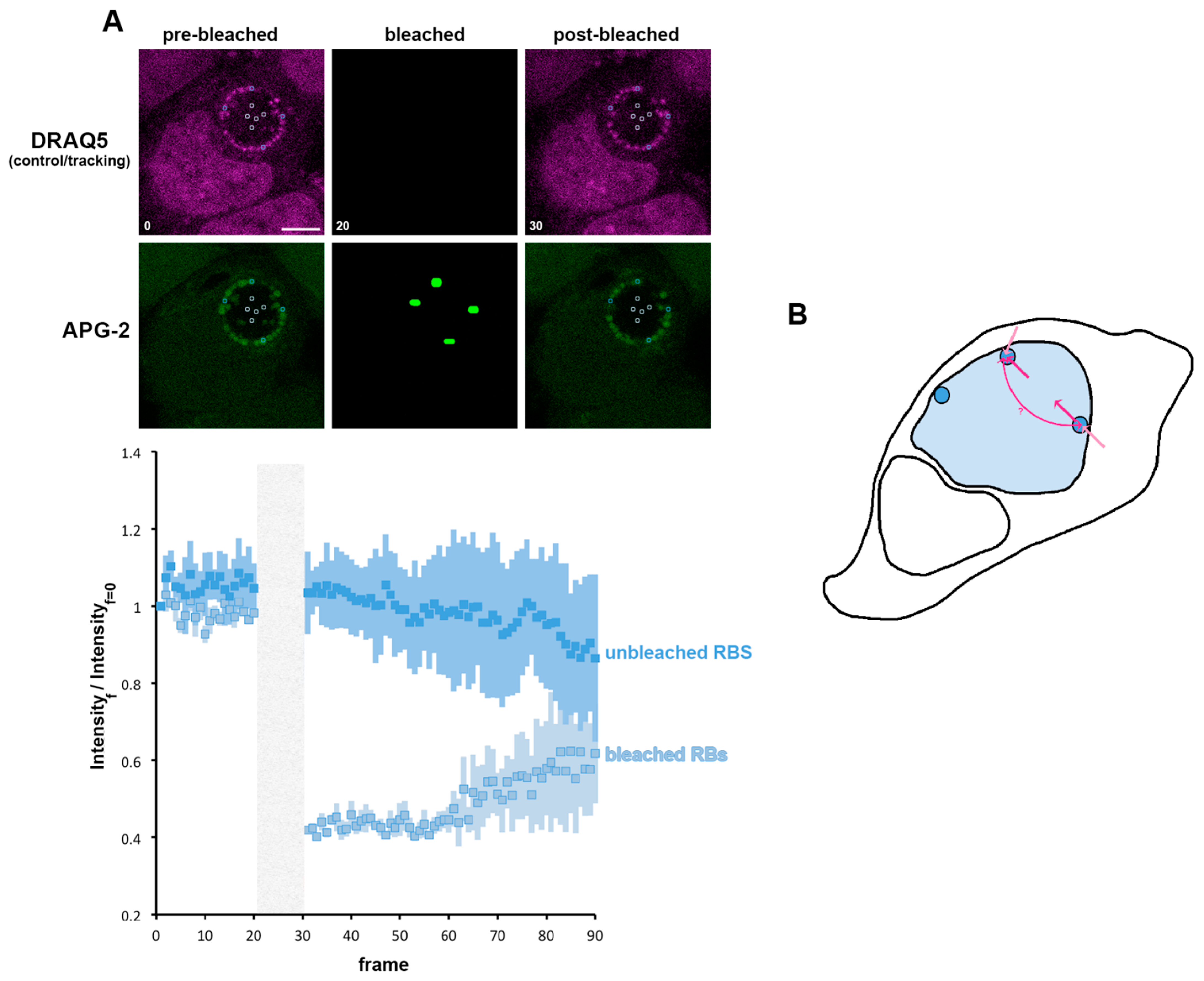
3.6. A Role for K+ in Bacterial Communication
3.7. A Role for K+ in Bacterial Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eley, A. Chlamydia trachomatis is bad for your sperm! Microbiol. Today 2003, 30, 61–62. [Google Scholar]
- Hogan, R.J.; Mathews, S.A.; Mukhopadhyay, S.; Summersgill, J.T.; Timms, P. Chlamydial Persistence: Beyond the Biphasic Paradigm. Infect. Immun. 2004, 72, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Mpiga, P.; Ravaoarinoro, M. Chlamydia trachomatis persistence: An update. Microbiol. Res. 2006, 161, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Skowasch, D.; Yeghiazaryan, K.; Schrempf, S.; Golubnitschaja, O.; Welsch, U.; Preusse, C.J.; A Likungu, J.; Welz, A.; Lüderitz, B.; Bauriedel, G. Persistence of Chlamydia pneumoniae in degenerative aortic valve stenosis indicated by heat shock protein 60 homologues. J. Heart Valve Dis. 2003, 12, 68–75. [Google Scholar] [PubMed]
- Borel, N.; Summersgill, J.T.; Mukhopadhyay, S.; Miller, R.D.; Ramirez, J.A.; Pospischil, A. Evidence for persistent Chlamydia pneumoniae infection of human coronary. Atherosclerosis 2008, 199, 154–161. [Google Scholar] [CrossRef]
- Beatty, W.L.; Morrison, R.P.; Byrne, G.I. Persistence in Chlamydiae: From cell culture to a paradigm for chlamydial pathogenesis. Microbiol. Rev. 1994, 58, 686–699. [Google Scholar] [CrossRef]
- Allan, I.; Pearce, J.H. Amino Acid Requirements of Strains of Chlamydia trachomatis and C. psittaci growing in McCoy Cells: Relationship with Clinical Syndrome and Host Origin. Microbiology 1983, 129, 2001–2007. [Google Scholar] [CrossRef]
- Shemer-Avni, Y.; Wallach, D.; Sarov, I. Inhibition of Chlamydia trachomatis growth by recombinant tumor necrosis factor. Infect. Immun. 1988, 56, 2503–2506. [Google Scholar] [CrossRef]
- Beatty, W.L.; Morrison, R.P.; I Byrne, G. Reactivation of persistent Chlamydia trachomatis infection in cell culture. Infect. Immun. 1995, 63, 199–205. [Google Scholar] [CrossRef]
- Pettengill, M.A.; Lam, V.W.; Ojcius, D.J. The Danger Signal Adenosine Induces Persistence of Chlamydial Infection through Stimulation of A2b Receptors. PLoS ONE 2009, 4, e8299. [Google Scholar] [CrossRef]
- Raulston, J.E. Response of Chlamydia trachomatis serovar E to iron restriction in vitro and evidence for iron-regulated chlamydial proteins. Infect. Immun. 1997, 65, 4539–4547. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.; Mirrashidi, K.; Engel, J.N. Chlamydia cell biology and pathogenesis. Nat. Rev. Genet. 2016, 14, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Enciso, G.A.; Boassa, D.; Chander, C.N.; Lou, T.H.; Pairawan, S.S.; Guo, M.C.; Wan, F.Y.M.; Ellisman, M.H.; Sütterlin, C.; et al. Replication-dependent size reduction precedes differentiation in Chlamydia trachomatis. Nat. Commun. 2018, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Prindle, A.; Liu, J.; Asally, M.; Ly, S.; Garcia-Ojalvo, J.; Süel, G.M. Ion channels enable electrical communication in bacterial communities. Nat. Cell Biol. 2015, 527, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. Boosting the signal: Endothelial inward rectifier K+ channels. Microcirculation 2017, 24, e12319. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Hollnagel, J.-O.; Elzoheiry, S.; Schneider, J. Energy and Potassium Ion Homeostasis during Gamma Oscillations. Front. Mol. Neurosci. 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Gong, H.; Lai, J.; Main, A.; Lu, S. The Potassium Transporter Trk and External Potassium Modulate Salmonella enterica Protein Secretion and Virulence. Infect. Immun. 2008, 77, 667–675. [Google Scholar] [CrossRef]
- Kumar, K.A.; Garcia, C.R.S.; Chandran, V.R.; Van Rooijen, N.V.; Zhou, Y.; Winzeler, E.; Nussenzweig, V. Exposure of Plasmodium sporozites to the intracellular concentration of potassium enhances infectivity and reduces cell passage activity. Mol. Biochem. Parasitol. 2007, 156, 32–40. [Google Scholar] [CrossRef]
- Moudy, R.; Manning, T.J.; Beckers, C.J. The Loss of Cytoplasmic Potassium upon Host Cell Breakdown Triggers Egress of Toxoplasma gondii. J. Biol. Chem. 2001, 276, 41492–41501. [Google Scholar] [CrossRef]
- Abdul-Sater, A.A.; Saïd-Sadier, N.; Padilla, E.V.; Ojcius, D.M. Chlamydial infection of monocytes stimulates IL-1b secretion through activation of the NLRP3 inflammasome. Microbes Infect. 2010, 12, 652–661. [Google Scholar] [CrossRef]
- Chang, G.T.; Moulder, J.W. Loss of inorganic ions from host cells infected with Chlamydia psittaci. Infect. Immun. 1978, 19, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Dumoux, M.; Clare, D.K.; Saibil, H.R.; Hayward, R.D. ChlamydiaeAssemble a Pathogen Synapse to Hijack the Host Endoplasmic Reticulum. Traffic 2012, 13, 1612–1627. [Google Scholar] [CrossRef] [PubMed]
- Wickstrum, J.; Sammons, L.R.; Restivo, K.N.; Hefty, P.S. Conditional Gene Expression in Chlamydia trachomatis using the Tet System. PLoS ONE 2013, 8, e76743. [Google Scholar] [CrossRef]
- Dumoux, M.; Le Gall, S.M.; Habbeddine, M.; Delarbre, C.; Hayward, R.D.; Kanellopoulos-Langevin, C.; Verbeke, P. Penicillin Kills Chlamydia following the Fusion of Bacteria with Lysosomes and Prevents Genital Inflammatory Lesions in C. muridarum-Infected Mice. PLoS ONE 2013, 8, e83511. [Google Scholar] [CrossRef]
- Smith, R.V.; Nessen, M.A. Atomic Absorption Analysis of Sodium, Potassium, and Calcium in Ringer’s Solution. J. Pharm. Sci. 1971, 60, 907–908. [Google Scholar] [CrossRef]
- Eliceiri, K.W.; Berthold, M.R.; Goldberg, I.G.; Ibáñez, L.; Manjunath, B.; Martone, M.E.; Murphy, R.F.; Peng, H.; Plant, A.L.; Roysam, B.; et al. Biological imaging software tools. Nat. Methods 2012, 9, 697–710. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Kappel, C.; Eils, R. Fluorescence recovery after photobleaching with the Leica TCS SP2. Confocal Appl. Lett. 2004, 18, 1–12. [Google Scholar]
- Shima, K.; Klinger, M.; Solbach, W.; Rupp, J. Activities of First-Choice Antimicrobials against Gamma Interferon-Treated Chlamydia trachomatis Differ in Hypoxia. Antimicrob. Agents Chemother. 2013, 57, 2828–2830. [Google Scholar] [CrossRef][Green Version]
- Barber, A. Intracellular free potassium, sodium and chloride measured with ion-selective microelectrodes from salivary gland cells of snail Planorbis Corneus. J. Exp. Biol. 1987, 128, 349–369. [Google Scholar]
- Nicholls, D.G.; Chou, C.-Y.; Jen, W.-P.; Hsieh, Y.-H.; Shiao, M.-S.; Chang, G.-G. Simultaneous Monitoring of Ionophore- and Inhibitor-mediated Plasma and Mitochondrial Membrane Potential Changes in Cultured Neurons. J. Biol. Chem. 2006, 281, 14864–14874. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bryan, L.; Bryan, J. Molecular Biology of Adenosine Triphosphate-Sensitive Potassium Channels. Endocr. Rev. 1999, 20, 101–135. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.J.; Baukrowitz, T. How Highly Charged Anionic Lipids Bind and Regulate Ion Channels. J. Gen. Physiol. 2008, 131, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Higashi, N. Electron microscopic studies on the mode of reproduction of trachoma virus and psittacosis virus in cell cultures. Exp. Mol. Pathol. 1965, 4, 24–39. [Google Scholar] [CrossRef]
- Wilson, D.P.; Whittum-Hudson, J.A.; Timms, P.; Bavoil, P.M. Kinematics of intracellular Chlamydiae provide evidence for contact-dependent development. J. Bacteriol. 2009, 191, 5734–5742. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, H.H.; Morré, D.J.; Rowe, L.D. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim. Biophys. Acta 1990, 1031, 225–246. [Google Scholar] [CrossRef]
- Gieffers, J.; Rupp, J.; Gebert, A.; Solbach, W.; Klinger, M. First-Choice Antibiotics at Subinhibitory Concentrations Induce Persistence of Chlamydia pneumoniae. Antimicrob. Agents Chemother. 2004, 48, 1402–1405. [Google Scholar] [CrossRef]
- Chiappino, M.L.; Dawson, C.; Schachter, J.; Nichols, B.A. Cytochemical localization of glycogen in Chlamydia trachomatis inclusions. J. Bacteriol. 1995, 177, 5358–5363. [Google Scholar] [CrossRef]
- Herzberg, M.; Britbart, H.; Atlan, H. Interaction between membrane functions and protein synthesis in reticulocytes. Eur. J. Biochem. 1974, 45, 161–170. [Google Scholar] [CrossRef]
- Gérard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P. Genes required for assembly and function of the protein syn-thetic system in Chlamydia trachomatis are expressed early in elementary to reticulate body transformation. Mol. Gen. Genet. 1997, 255, 637–642. [Google Scholar] [CrossRef]
- Sugi, K.; Mush, M.W.; Field, M.; Chang, E.B. Inhibition of Na+, K+-ATPase by interferon gamma down-regulates in-testinal epithelial transport and barrier function. Gastroenterology 2001, 120, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Atienza, F.; Almendral, J.; Moreno, J.; Vaidyanathan, R.; Talkachou, A.; Kalifa, J.; Arenal, A.; Villacastin, J.P.; Torrecilla, E.G.; Sanchez, A.; et al. Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: Evidence for a re-entrant mechanism. Circulation 2006, 114, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hosoya, Y.; Inanobe, A.; Tomoike, H.; Endoh, M. Acetylcholine and adenosine activate the G protein-gated muscarinic K+ channel in ferret ventricular myocytes. Naunyn Schmiedeberg’s Arch. Pharmacol. 1995, 351, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Krulwich, T.A.; Sachs, G.; Padan, E. Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Genet. 2011, 9, 330–343. [Google Scholar] [CrossRef]
- Grieshaber, S.; Swanson, J.; Hackstadt, T. Determination of the physical environment within the Chlamydia trachomatis inclusion using ion-selective ratiometric probes. Cell. Microbiol. 2002, 4, 273–283. [Google Scholar] [CrossRef]
- Kem, D.C.; Trachewsky, D. Potassium metabolism. In Potassium: Its Biologic Significances; CRC Press: Boca Raton, FL, USA, 1983; pp. 25–35. [Google Scholar]
- Whatmore, A.M.; Reed, R.H. Determination of turgor pressure in Bacillus subtilis: A possible role for K+ in turgor reg-ulation. J. Gen. Microbiol. 1990, 136, 2521–2526. [Google Scholar] [CrossRef]
- Epstein, W. The Roles and Regulation of Potassium in Bacteria. Prog. Nucleic Acid Res. Mol. Biol. 2003, 75, 293–320. [Google Scholar]
- Comes, N.; Serrano-Albarrás, A.; Capera, J.; Serrano-Novillo, C.; Condom, E.; Cajal, S.R.Y.; Ferreres, J.C.; Felipe, A. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2477–2492. [Google Scholar] [CrossRef]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 1966, 64, 445–502. [Google Scholar] [CrossRef]
- Harold, F.M. Membranes and Energy Transduction in Bacteria. Curr. Top. Bioenerg. 1977, 6, 83–149. [Google Scholar]
- Dean, D.; Myers, G.S.; Read, T.D. Lessons and challenges arising from the ‘first wave’ of Chlamydia genome sequencing. In Chlamydia: Genomics and Pathogenesis; Horizon Scientific Press: Poole, UK, 2006. [Google Scholar]
- Ashraf, K.U.; Josts, I.; Mosbahi, K.; Kelly, S.M.; Byron, O.; Smith, B.O.; Walker, D. The potassium binding protein KBP is a cytoplasmic potassium sensor. Structure 2016, 24, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Strahl, H.; Hamoen, L.W. Membrane potential is important for bacterial cell division. Proc. Natl. Acad. Sci. USA 2010, 107, 12281–12286. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrew, S.C.; Dumoux, M.; Hayward, R.D. Chlamydia Uses K+ Electrical Signalling to Orchestrate Host Sensing, Inter-Bacterial Communication and Differentiation. Microorganisms 2021, 9, 173. https://doi.org/10.3390/microorganisms9010173
Andrew SC, Dumoux M, Hayward RD. Chlamydia Uses K+ Electrical Signalling to Orchestrate Host Sensing, Inter-Bacterial Communication and Differentiation. Microorganisms. 2021; 9(1):173. https://doi.org/10.3390/microorganisms9010173
Chicago/Turabian StyleAndrew, Susan C., Maud Dumoux, and Richard D. Hayward. 2021. "Chlamydia Uses K+ Electrical Signalling to Orchestrate Host Sensing, Inter-Bacterial Communication and Differentiation" Microorganisms 9, no. 1: 173. https://doi.org/10.3390/microorganisms9010173
APA StyleAndrew, S. C., Dumoux, M., & Hayward, R. D. (2021). Chlamydia Uses K+ Electrical Signalling to Orchestrate Host Sensing, Inter-Bacterial Communication and Differentiation. Microorganisms, 9(1), 173. https://doi.org/10.3390/microorganisms9010173