1. Introduction
Polyphosphates (polyP) are polymers of inorganic orthophosphate that can reach hundreds to thousands of residues in size. PPK and PPX are the two main enzymes related to polyP metabolism in Prokaryotes. PPK synthesizes polyP, consuming ATP as the polyP chain grows, generating ADP and polyP (n+1) as final products. On the other hand, PPX degrades polyP starting from its last phosphate group, releasing Pi and polyP (n−1) in the process [
1]. In archaea, PPX has been identified either by experimental [
2] or bioinformatic analysis [
3]. Nevertheless, even though polyP accumulates in some Crenarchaeotes, no archaeal PPK homolog has been found so far, suggesting a different enzyme involved in polyP synthesis in this phylum.
Many functions of polyP have been revealed in bacteria such as: ATP substitution, helping in nutrition starvation, functioning in metal resistance, and several other roles [
1,
4]. Some of those functions are related to biofilm formation, motility, and sporulation [
5,
6,
7,
8] as shown with several PPK and/or PPX mutants in diverse bacterial species.
A
ppk deletion mutant in
Pseudomonas aeruginosa showed a thinner and more uniform biofilm when compared to the wild type (WT) strain and lacked cells forming clusters along with water channels [
6]. This mutant was defective in swimming, swarming, twitching and adhesion to surfaces. All of these phenomena are related to biofilm formation. Involvement of polyP in biofilm development, sporulation and even virulence was also shown in
Bacillus cereus [
7] and
Campylobacter jejuni [
5].
In
E. coli, the role of polyP in biofilm generation has been studied in more detail. Rather than the intracellular level of the polymer, polyP degradation triggers biofilm formation in stationary phase via Type 2 Autoinducers (AI-2), specifically the LuxS Quorum Sensing system [
9]. A correlation between polyP levels and AI-1 and AI-2 was also seen in
P. aeruginosa [
6].
The possible role of polyP in biofilm formation in archaea has not been explored. Archaea lack AI-2, suggesting that regulation in these microorganisms functions differently. Regulation of biofilm formation in
Sulfolobus acidocaldarius involves two transcriptional regulators belonging to the Lrs-14 family: encoded by Saci_1223 and Saci_0446, the latter is known as AbfR1 (Archaeal Biofilm Regulator 1) [
10]. Saci_1223 is a positive regulator, inducing biofilm formation, whereas AbfR1 is a negative regulator that inhibits biofilm development by repressing extracellular polymeric substances (EPS) synthesis and promoting swimming motility by using archaella. AbfR1 is regulated by phosphorylation [
11] and cannot impair biofilm formation when phosphorylated. The pathway for EPS synthesis has not been described yet for
Sulfolobales.
Here we show that polyP is also related to biofilm formation in archaea, affecting both attachment of cells to surfaces and motility, apparently through assembly of the archaellum (archaeal motility structure), in two model crenarchaeotes Sa. solfataricus and S. acidocaldarius.
2. Materials and Methods
2.1. Strains and Growth Conditions
Sa. solfataricus M16 and its polyP (−) strain [
12], as well as
S. acidocaldarius MW001 and its corresponding polyP (−) strain were grown as before at 75 °C by shaking at 150 rpm in Brock Medium [
12], pH 3 and supplemented with 0.1% (
p/v) N-Z amine (Sigma-Aldrich
®, Merk KGaA, Burlington, MA, USA), 0.2% (
p/v) glucose and 0.01 mg/mL uracil only in the case of uracil autotrophs. For overexpression of PPX (described below), 0.2% (
w/v)
d-arabinose (Sigma-Aldrich
®, Merk KGaA, Burlington, MA, USA) was added.
2.2. Construction of a S. acidocaldarius Mutant Lacking PolyP
To generate recombinant
S. acidocaldarius, the
ppx gene (Saci_2018) was amplified from
S. acidocaldarius MW001 genomic DNA using PCR with primers 10904 and 10905 (
Table S2). The obtained product was cloned into pSVAaraFX-H6 [
13] using
SapI (New England Biolabs., Ipswich, MA, USA), resulting in pSVA12801. The plasmid was sequenced.
Before being introduced in
S. acidocaldarius MW001, the plasmid was methylated by transformation and purification from
E. coli ER1821 strain. Methylated plasmids were transformed to competent
S. acidocaldarius MW001 as described in [
14] and plated on Brock first selection plates. Colonies were picked and grown in Brock medium supplemented with N-Z amine and Glucose.
The overexpression of PPX enzyme was induced for 3 h with 0.2% d-arabinose while growing in liquid Brock medium with supplements. Cells were collected by centrifugation and boiled in protein buffer (0.5 M Tris-HCl, pH 6.8, 65 mM glycerol, 10% SDS, 2% 2-mercapto ethanol, 0.025% bromophenol blue) at 100 °C for 5 min.
2.3. PolyP Extraction and Quantification
Cultures of
S. acidocaldarius were grown as described in
Section 2.1. PolyP from
S. acidocaldarius cells were isolated as previously described [
12]. In brief, 5 mL culture was centrifuged for 15 min at 4500×
g. Cell pellet was resuspended in prewarmed 300 μL of 4 M guanidine isothiocyanate (GITC), 50 mM Tris-HCl pH 7.0 solution. The suspension was subjected to vortex and heated for 3 min at 95 °C. Twenty µL of each sample was saved for protein quantification using Coomassie Plus Protein Assay Reagent (PIERCE™, Thermo Fisher Scientific, Waltham, MA, USA). Thirty µL of 10% SDS was added to each sample and then heated again at 95 °C for 5 min. To bind polyP, 5 µL of silica (Glassmilk) and 300 µL of ethanol (100%) were added and mixed by vortexing followed by heating at 95 °C for 30 s. After 1 min centrifugation at 13,000×
g, the supernatant was removed and the glassmilk pellet was resuspended in 200 µL of ice cool New Wash Buffer (5 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM EDTA, 50% ethanol). After centrifugation, glassmilk was resuspended in 100 µL of a solution containing 50 mM Tris-HCl pH 7.0, 5 mM MgCl
2, 5 µg/mL DNase and 5 µg/mL RNase and incubated for 30 min at 37 °C to eliminate nucleic acids. The pellet was washed twice with New Wash Buffer. Finally, a total of 100 µL of polyP-containing solution was obtained and polyP was recovered from the glassmilk after repeatedly vortexing with 50 µL of water, heating at 95 °C and centrifugation. If not measured immediately, samples were frozen and stored at −20 °C.
Quantification of total Pi was done with EnzChek Phosphate Assay kit (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s instructions and expressed as nmol of Pi per mg of protein. Thirty µL of polyP containing solution was subjected to acid hydrolysis with 30 µL 2 N HCl for 30 min at 95 °C to release inorganic phosphate (Pi), followed by quantification.
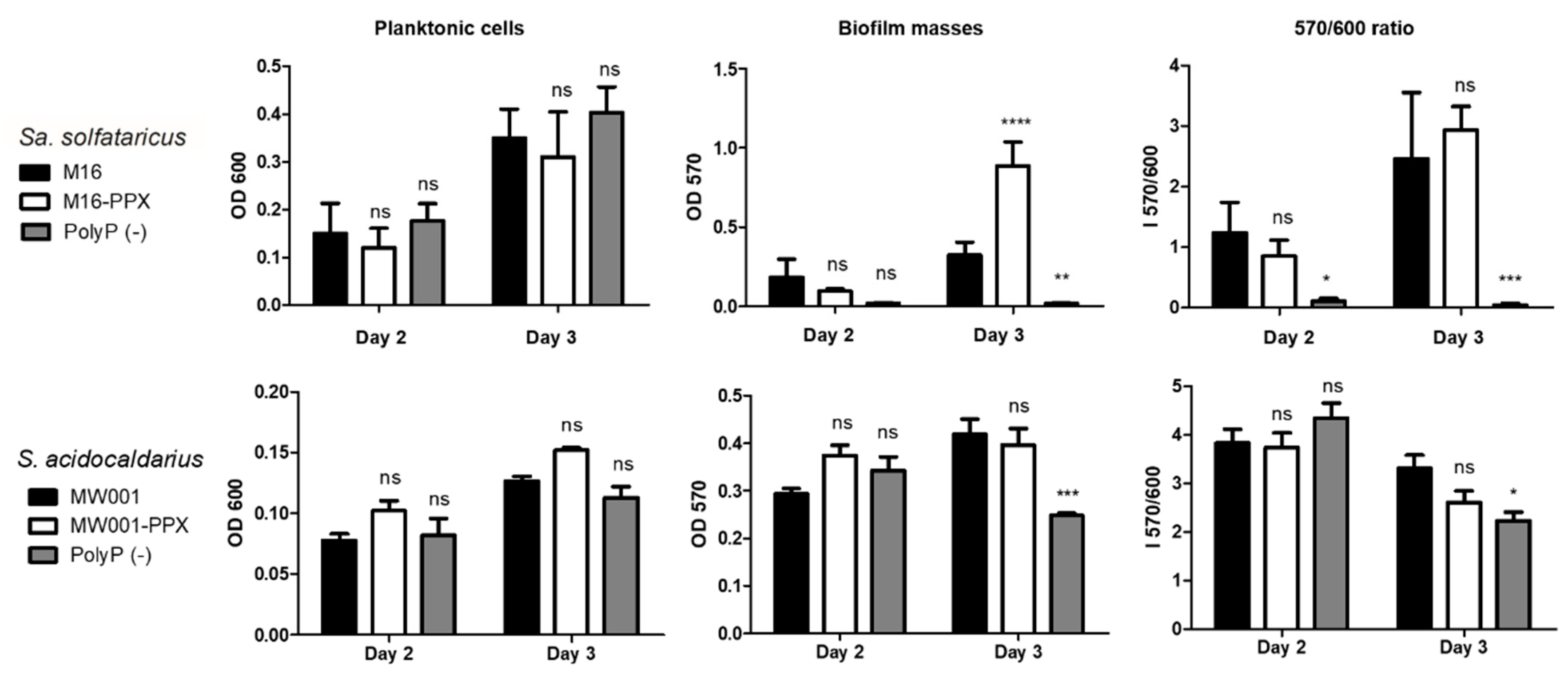
2.4. Microtitration Plates Assays
Cells were grown to stationary phase in Brock medium (pH 3.5) supplemented with 0.1% N-Z-amine and 0.2%
d-arabinose or uracil in 96 wells microtitration plates covered with gas permeable sealing membrane (Breathe-Easy, Diversified Biotech, Boston, MA, USA) at 75 °C for 2 or 3 days inside a humidity chamber to avoid evaporation. The starting OD
600 was 0.03 for
Sa. solfataricus and 0.01 for
S. acidocaldarius, as previously described by Koerdt et al. [
15]. After 2 or 3 days, plates were cooled down, the supernatant was placed in a new plate and the OD
600 was determined by an Epoch luminometer (BioTek instruments, Agilent Technologies, Inc., Santa Clara, USA). Ten µL of 0.5% crystal violet solution (CV) was added to each well containing the biofilm followed by incubation for 10 min. Sessile cells were washed with Brock (pH 5) medium or water, and CV attached to the biofilm was released by using 200 µL of 30% acetic acid. The CV released was determined by measuring the OD
570. The OD
570/OD
600 correlation index was used to determine biofilm formation efficiency.
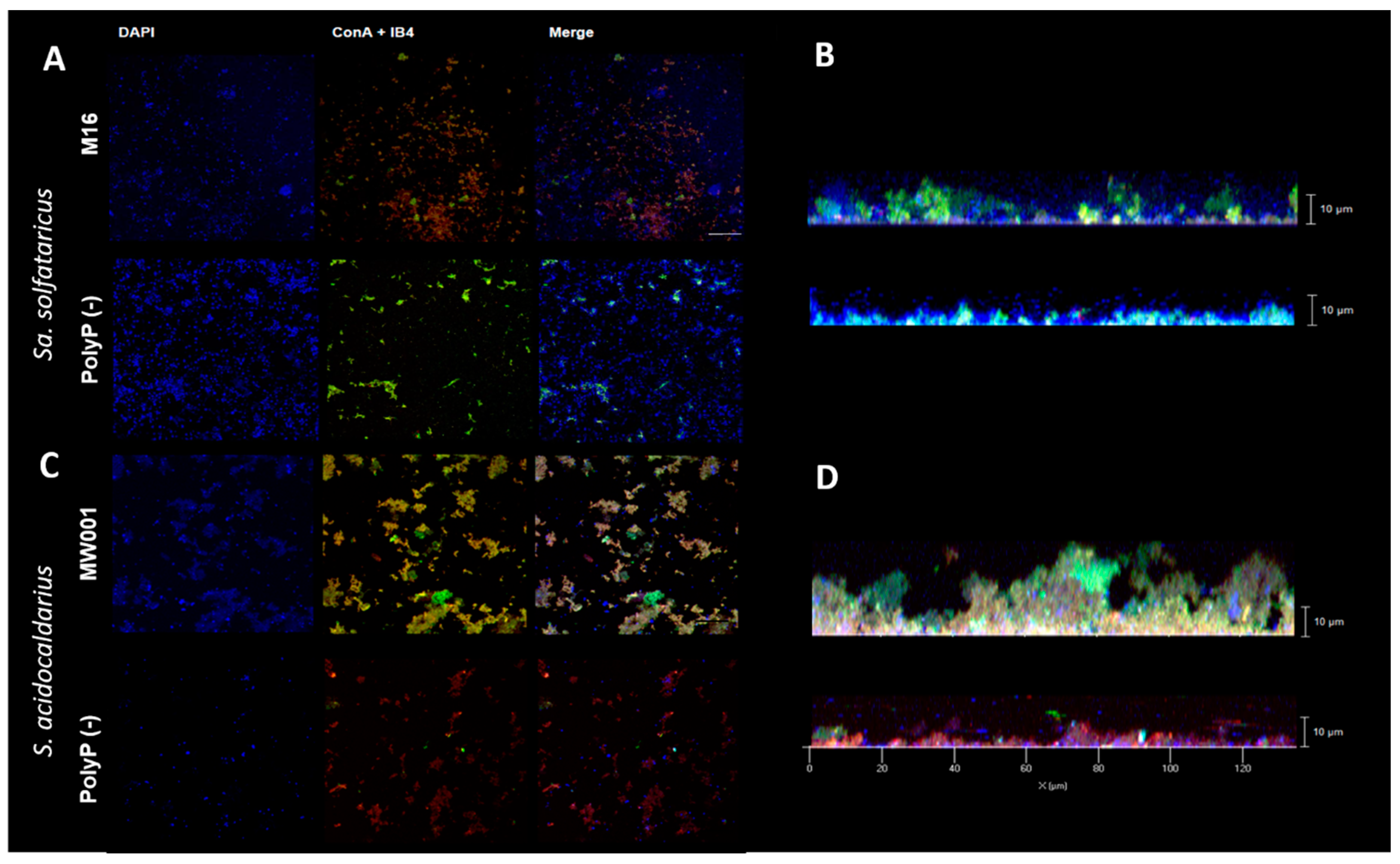
2.5. Confocal Laser Microscopy
For Confocal Laser Microscopy (CLM), cells were grown in 35 mm petri dishes (µ-dishes; Ibidi; Martinsried) [
15]. Culture medium was exchanged every 24 h, and after 3 days, the supernatant was exchanged for 2 mL Brock (pH 5) and biofilm was stained with 3.6 µL of DAPI (4′,6-diamidino-2-phenylindole) (500 µg/mL), 15 µL ConA-fluoresceine (5 mg/mL) and IB4-Alexa568 (lectin IB4 from
Griffonia simplicifolia, Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) was used for CLM. Staining was done for 30 min in the dark at room temperature.
Biofilms were observed under microscope ZEISS Observer 1 with 63× objective
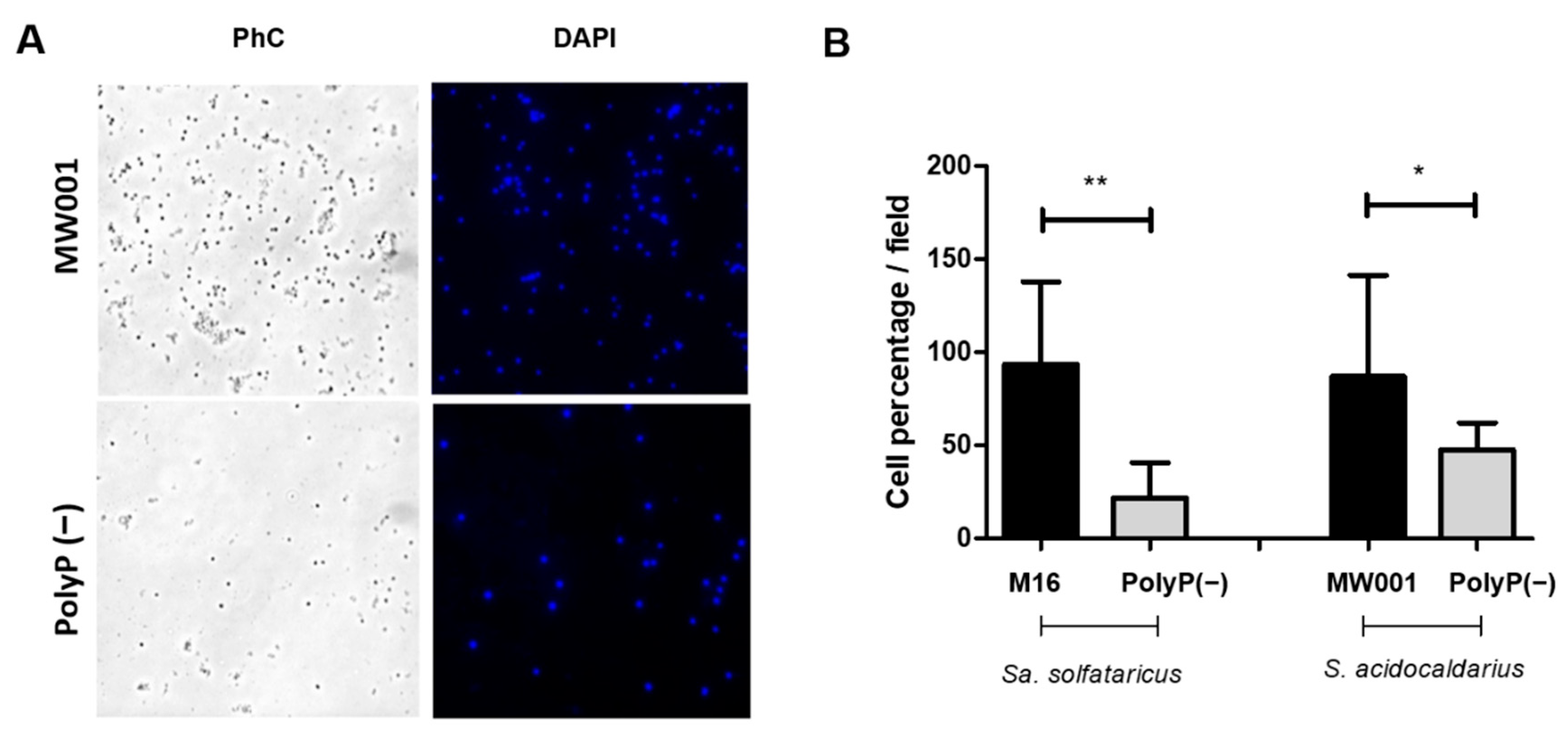
2.6. Adhesion Assays
For adhesion assays, 40 mL of
Sa. solfataricus or
S. acidocaldarius cultures with an initial OD
600 of 0.03 and 0.01, respectively, were grown to OD
600 of 0.5–0.7 for 24 h at 75 °C and 150 rpm in a 100 mL Schott flask with a glass slide inside. After that period, the glass slide was removed, washed twice with Brock medium (pH 5), and cells attached were fixed with 4% formaldehyde dissolved in Brock medium (pH 5). Cells and EPS were stained with 6 µL DAPI (300 µg/mL) and 15 µL (5 mg/mL) ConA dissolved in 1 mL Brock medium (pH 5) for 30 min in the dark. After staining, slides were washed twice with Brock medium and air dried. Cells in the back part of the slide were removed with 70% ethanol. Slides were observed by using the TIRF Observer 1 from ZEISS microscope with a 100× objective. Ten pictures from different fields were taken from each of 3 biological replicates and processed with Fiji (ImageJ) [
16].
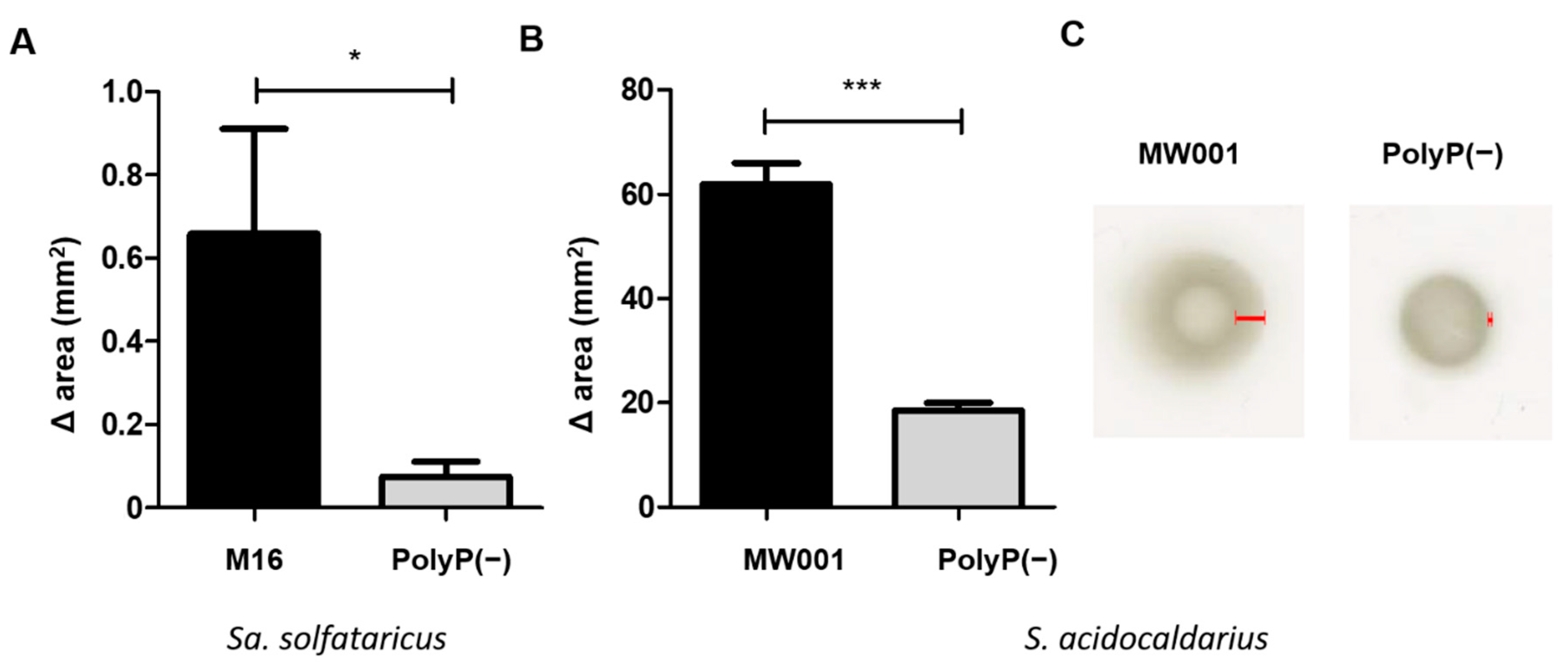
2.7. Motility Assays
Motility was analyzed on semi-solid gelrite plates. Motility plates were made with Brock medium pH 5, 0.15% gelrite and supplemented with 0.0001% N-Z amine, 0.2% d-arabinose and uracil when needed. Cells for inoculation were grown to an OD600 between 0.4 and 0.6, placed as a spot on the surface of the motility plates and air dried. Plates were incubated for 4 days in the case of S. acidocaldarius strains, and 9 days for Sa. solfataricus strains inside a humidity chamber. Finally, the swimming radius/area of each spot was measured.
2.8. Total RNA Extraction and cDNA Synthesis
To study the expression of genes of interest, cells were grown in large 150 mm petri dishes for biofilm formation during 3 days at 75 °C and no agitation inside a humidity chamber, exchanging the medium after the first 24 h. Cells were grown in Brock medium, starting at an initial OD600 of 0.03. For polyP (−) strains, d-arabinose was added. For planktonic cells, cultures were grown in the same conditions but in Erlenmeyer flasks.
After 3 days, the supernatant from the biofilms was removed and cells from the biofilm were washed twice with Brock medium and scraped from the bottom with a cell scraper and 20 mL of fresh medium. Biofilm and planktonic cells (10 mg wet weight) were harvested by centrifugation (7700×
g for 15 min). Cell pellets were washed three times with Brock’s medium and lysed as previously described [
17]. RNA was extracted by using TRIzol (Invitrogen™, Thermo Fisher Scientific, Waltham, MA USA) as described by the manufacturer. Remaining DNA was eliminated by adding 40 U of TURBO DNA-free DNase (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s instructions. 0.8 µg of total RNA was reverse transcribed for cDNA synthesis using ImProm-II (Promega, Madison, WI, USA), 0.5 µg of random hexamers (Promega, Madison, WI, USA) and 3 mM MgCl
2 for 1 h at 42 °C. Three biological replicates were used for every experimental condition.
2.9. Primer Design and Real-Time RT-PCR
Primers for qRT-PCR were designed using Primer3 Software and the annotated genome of
Sa. solfataricus P2
and S. acidocaldarius DSM639, as they are the WT strains [
14,
18] (
Table S1). All primers are listed in
Table S2. To check primer specific annealing and optimal melting temperature, PCR reactions were carried out with Taq DNA polymerase from Promega following manufacturer’s instructions and the products were separated by gel electrophoresis (1% agarose).
Gene expression was analyzed with the 96-well PikoReal-Time PCR System (Thermo Fisher Scientific, Waltham, USA). Five µL of KAPA SYBR® FAST 2X (Sigma-Aldrich®, Merk KGaA, Burlington, USA) was used along 0.2 µL of each primer and 0.5 µL of a 1:20 dilution of the cDNA.
The efficiency of each pair of primers was calculated from the average slope of a linear regression curve, constructed from qPCRs using a 10-fold dilution series (10 pg–10 ng) of Sa. solfataricus M16 or S. acidocaldarius MW001 chromosomal DNA as template. Cq values (quantification cycle) after 40 cycles were automatically determined by PikoReal Software 2.1 (Thermo Fisher Scientific, Waltham, MA, USA). Cq values of each transcript of interest was standardized to the Cq value of the 16s and/or 30s rRNA gene. At least 3 biological replicates of each assessed condition and 2 technical replicates per qPCR reaction were performed. Rps2P and 16S rRNA were used as housekeeping genes.
2.10. Starvation Induction Experiment
S. acidocaldarius MW001 and polyP (−) were grown in Brock medium and supplemented with 0.1% (p/v) N-Z amine (Sigma-Aldrich®, Merk KGaA, Burlington, USA), 0.2% (p/v) dextrose and 0.01 mg/mL uracil only in the case of S. acidocaldarius MW001. For overexpression of PPX, 0.2% (w/v) d-arabinose (Sigma-Aldrich®, Merk KGaA, Burlington, USA) was added. A control of S. acidocaldarius MW001 with plasmid pSVA12801 without induction was used.
After reaching an OD600 of 0.4–0.5, cells were centrifuged for 10 min at 4400 rpm and 70 °C and resuspended in starvation medium without supplements, but with uracil and d-arabinose when needed. Cells were grown for 4 h at 75 °C and 150 rpm, and then used for Transmission Electron Microscopy.
2.11. Transmission Electron Microscopy
Five µL of cells from the starvation experiment were applied on a glow-discharged 300 mesh formvar and carbon-coated copper grid (Plano GmbH, Wetzlar, Germany) and incubated for 10 s. The excess liquid was blotted away and the application step was repeated 8 times subsequently. Afterwards cells were stained with 2% (w/v) uranyl acetate. Imaging was performed using Zeiss Leo 912 Omega (Tungsten) (Carl Zeiss, Oberkochen, Germany) operated at 80 kV. Images were taken with Dual Speed 2K-On-Axis charged-coupled device (CCD) camera TRS, Sharp-Eye (TRS Systems, Moorenweis, Germany).
2.12. Statistical Analysis
Data obtained was subjected to analysis of variance (ANOVA) and a Bonferroni’s test in the Prism 8 GraphPad software.
4. Discussion
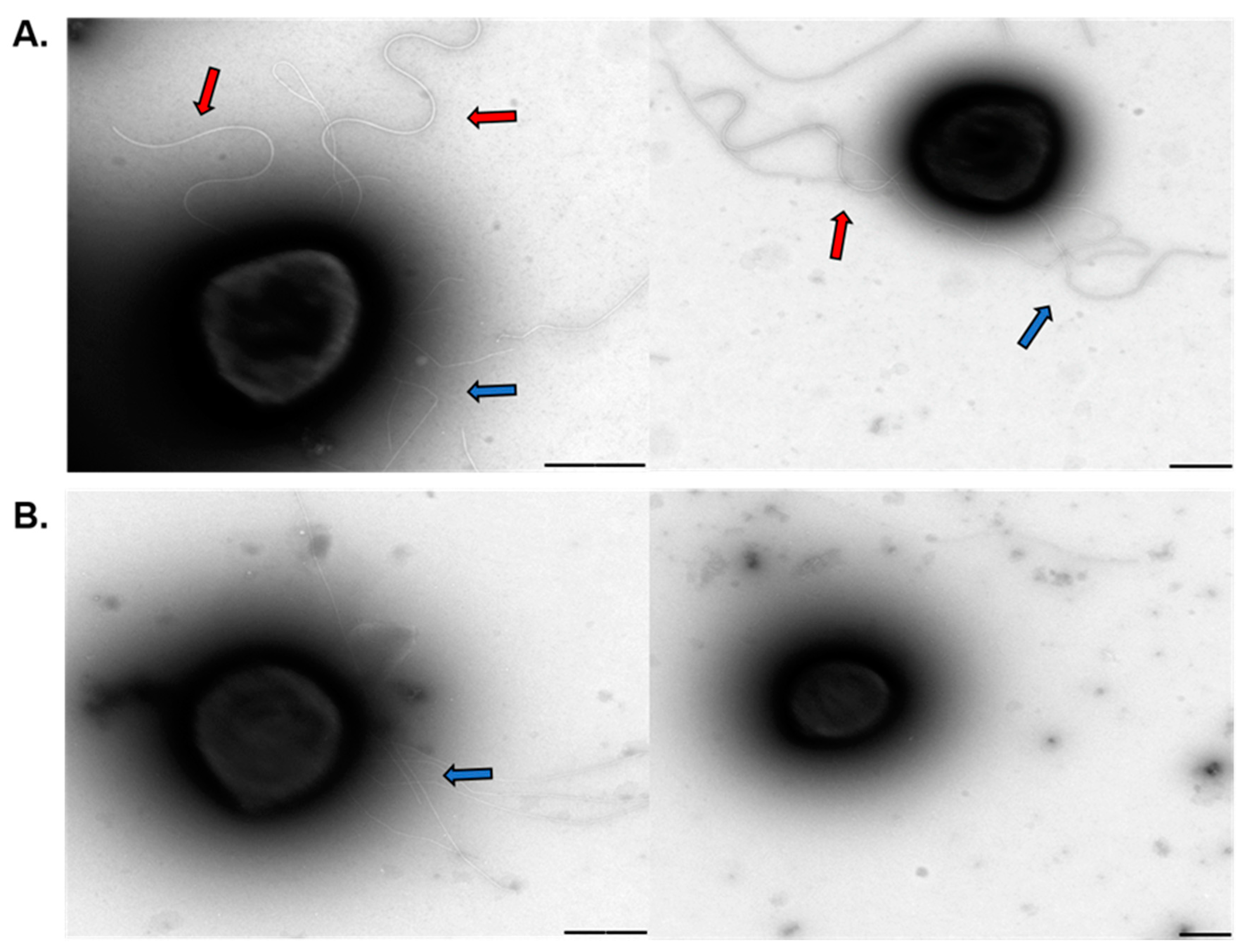
Overexpression of the ppx gene in both Sa. solfataricus and S. acidocaldarius eliminates polyP from their cells. Therefore, the use of these overexpression strains allowed to study the effect of polyP on biofilm formation, adhesion, and motility in archaeal cells.
Similar to what was observed for cells lacking the archaellum [
19,
21], polyP (−) cells exhibited a reduced surface-attachment and were not motile. Moreover, the
S. acidocaldarius polyP (−) strain produced a compact biofilm, with less amounts of EPS as observed for the Aap-pilus deletion mutant and the double deletion mutant for archaellum and Aap-pilus [
21]. The results presented here therefore suggest that similar to what is known in bacteria, polyP in
Sulfolobales functions in biofilm formation and motility by regulating the production of cell-surface structures that are known to be involved in the initial cellular swimming and attachment to surfaces [
19,
20,
21].
The exact mechanism of how polyP regulates biofilm formation is not known. In
E. coli, the degradation of the polymer at the beginning of stationary phase is related to LuxS and the formation of biofilm [
9]. In
Sulfolobales, no Quorum sensing system is known. Therefore, polyP must regulate biofilm formation in a different way.
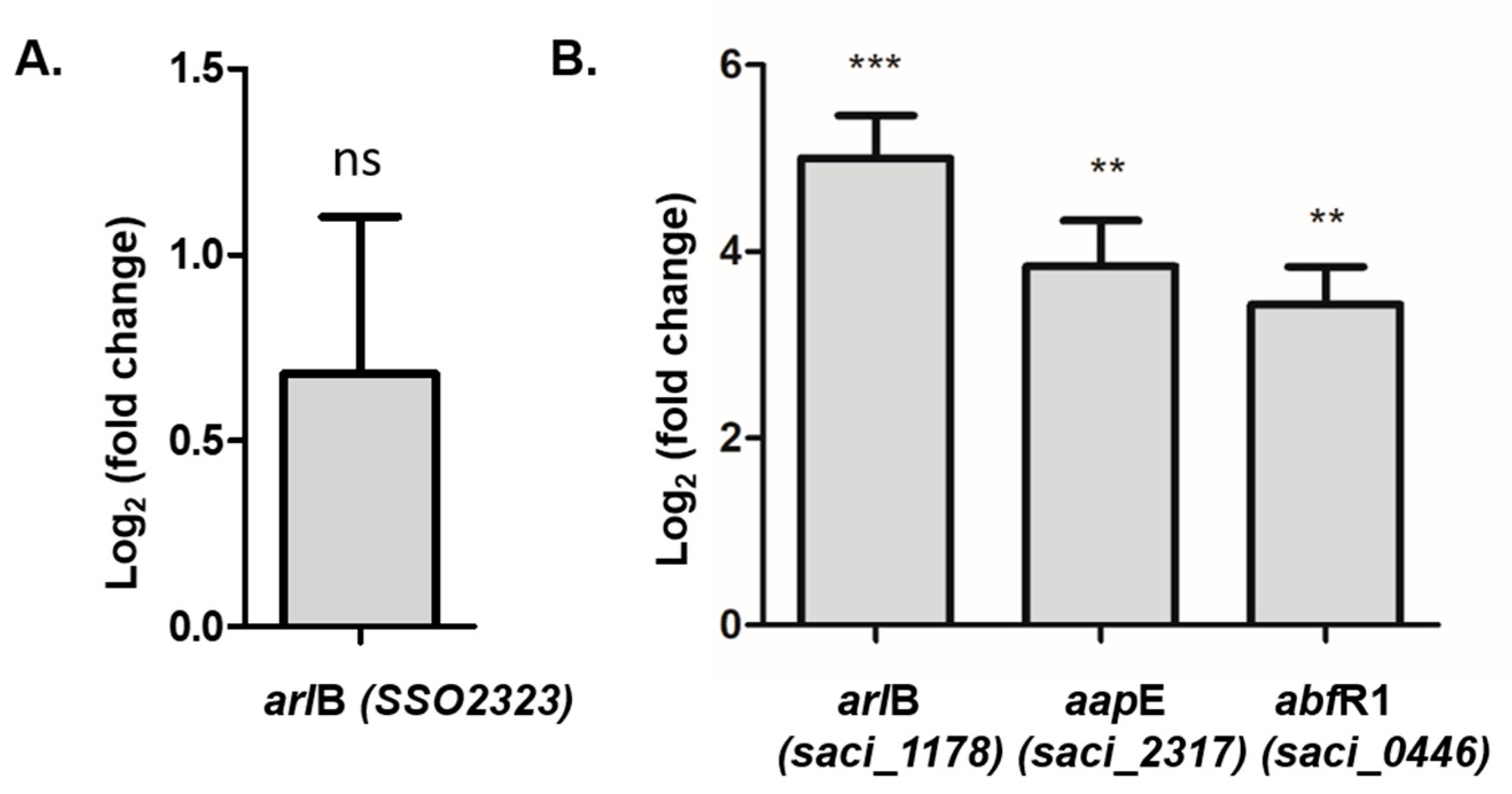
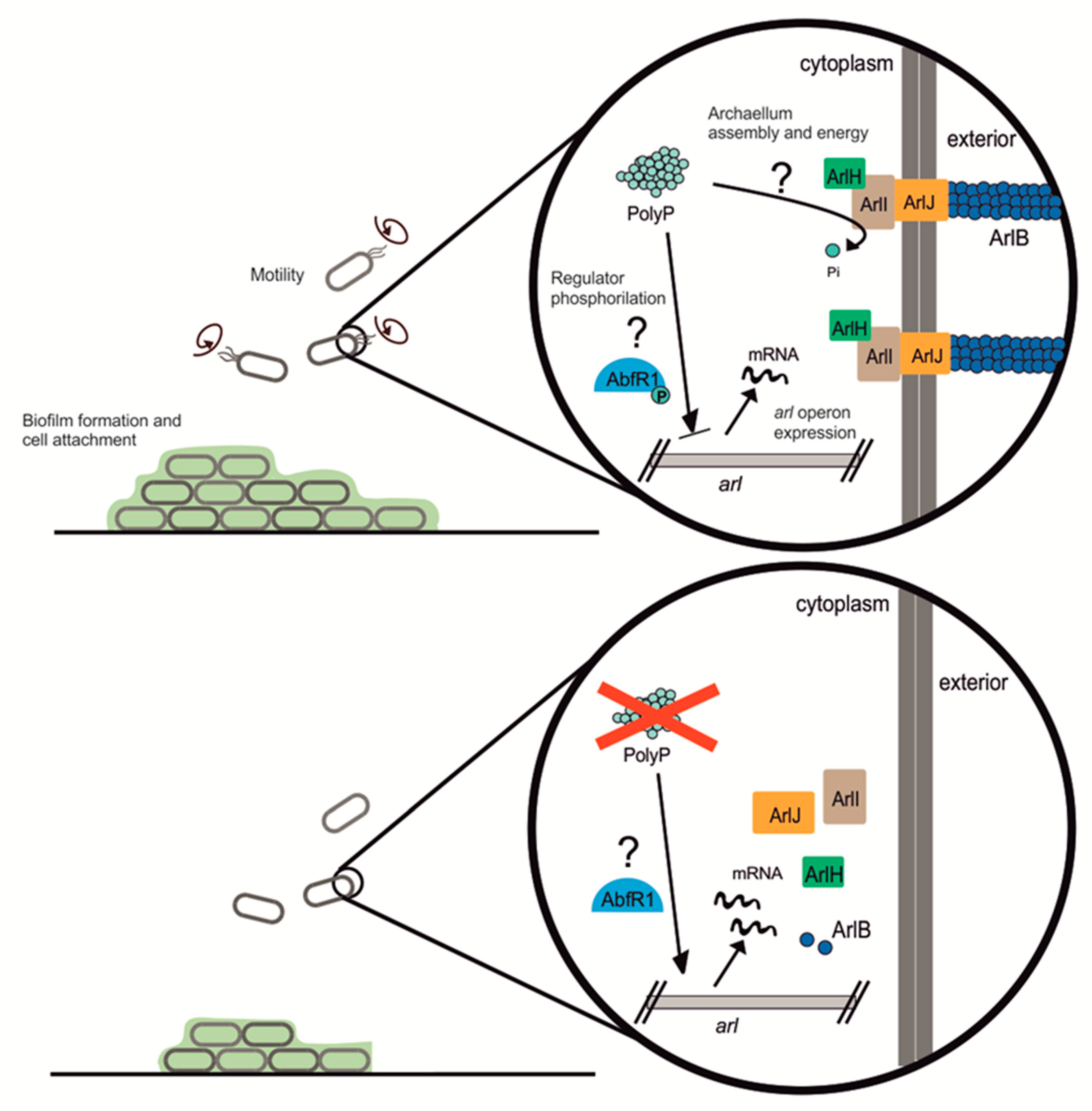
S. acidocaldarius contains two main regulators from the Lrs-14 family: Saci_1223 and AbfR1 [
10]. AbfR1 was of particular interest since it is itself regulated by phosphorylation. AbfR1 acts by inhibiting biofilm formation via induction of the
arl operon and reduction of EPS synthesis [
11] (see
Figure 7). AbfR1 also induces its own expression. However, when phosphorylated, AbfR1 cannot bind its regulation targets (
arlB promoter and its own
abfR1 promoter regions). Here we found that
abfR1 mRNA levels are higher in the polyP (−) strain, which explains in part the lower amount of biofilm formation. A possible explanation to this phenomenon is that the lack of polyP affects the regulation of AbfR1 by impairing phosphorylation, which would lead not only to the over-expression of AbfR1 itself but also the overexpression of the
arl operon (
Figure 7).
Deficient protein phosphorylation-disturbing expression of the archaellum genes was seen before for
S. acidocaldarius [
24], where cells lacking protein phosphatases genes (Saci_0545 and Saci_0884) showed overexpression of archaellum but less motility on semi-solid plates compared with the WT counterpart. However,
Sa. solfataricus AbfR1 (SSO_0458) does not harbor the amino acids proposed to be phosphorylated in
S. acidocaldarius (Y84 and S87) [
11]. It is therefore unclear how polyP is affecting phosphorylation of this regulator in both species. It is possible that polyP might also function in providing energy for assembly of the archaellum and Aap-pili [
22]. The assembly of the archaellum is energized by ATP hydrolysis [
25]. In
E. coli ppk and
ppx knockout mutants had lower expression of genes coding for fimbria and flagellum and were not flagellated as seen under the microscope [
26]. Although
Pseudomonas sp. B4 polyP (−) mutant showed lower levels of flagellin, intact flagella were also seen at its surface [
8].
It is interesting to consider that ppx-gene overexpression could generate an artificial ATP-consuming futile cycle in the cells, which is expected to lower the availability of ATP and thus the general energy load of the microorganisms. In future studies, it will be of importance to consider this possible effect, especially since deprivation of ATP might also affect archaella assembly and its movements. In addition, measuring the effects on cellular ATP concentration or heat generation (by microcalorimetrically), or on the relative fitness of the cells, would also be of interest in potential studies.
It is possible that an eventual lack of energy might take place in both
Sa. solfataricus and
S. acidocaldarius strains that do not accumulate polyP due to the overexpression of their respective PPX enzymes. This might create a futile cycle due to the constant polyP degradation, which in turn can cause an eventual lack of energy in these Crenarchaeotes. This situation might in turn explain the lack of motility and low biofilm formation phenotypes reported in this work. It is known that both of these strains normally accumulate low levels of polyP in their cytoplasm [
27]. Consequently, the amount of ATP required for the polymer synthesis should not be high. A bacterial kind of PPK type has not yet been identified in
Sulfolobales. In addition,
Sulfolobus species possess an ATP synthase enzymatic complex able to use the proton motive force to regenerate ATP [
28]. Considering these species grow at acid pH, ATP production could be favored in a natural way under their growth conditions. Each phosphate extension of the polyP polymer spends one ATP molecule and generates one of ADP. On the other hand, polyP degradation by the PPX enzyme would release one molecule of inorganic phosphate by each broken phosphodiester bond. Given these facts, one can consider that these archaeal strains could use both ADP and Pi substrates together with the energy of proton motive force to regenerate ATP from both polyP synthesis and degradation.
5. Conclusions
Even though polyP plays many functions in both eukaryotic and prokaryotic species, only little is known about the role of polyP in archaea. Here we report that polyP is important in biofilm formation, adhesion, motility, expression, and assembly of the archaellum in crenarchaeotes Sa. solfataricus and S. acidocaldarius. polyP (−) cells were not archaellated and showed less pili compared to the background strains used in this study. The lack of archaellum and pili in cell surface explains the lower motility and adhesion to glass surface, as well as some of the characteristics of the biofilm structure in these strains. We propose polyP might be affecting phosphorylation of proteins such as AbfR1, which is involved in biofilm formation as well as expression of Arl operon. At the same time, lack of polyP affects the assembly of the surface structures. Other levels of regulation should not be discarded meanwhile this study opens new questions about different roles of polyP in archaea.
This is the first evidence that polyP influences the mentioned phenomena in Archaea. By using genetically tractable archaea such as S. acidocaldarius, future studies would help us to determine the exact mode of regulation of archaeal polyP.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






