The Evolutionary Dynamics of Influenza A Viruses Circulating in Mallards in Duck Hunting Preserves in Maryland, USA

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Laboratory Diagnosis
2.2. Viral RNA Extraction, Whole-Genome Sequencing, Assembly and Annotation
2.3. Phylogenetic Analysis
3. Results
3.1. IAVs Detected in DHPs
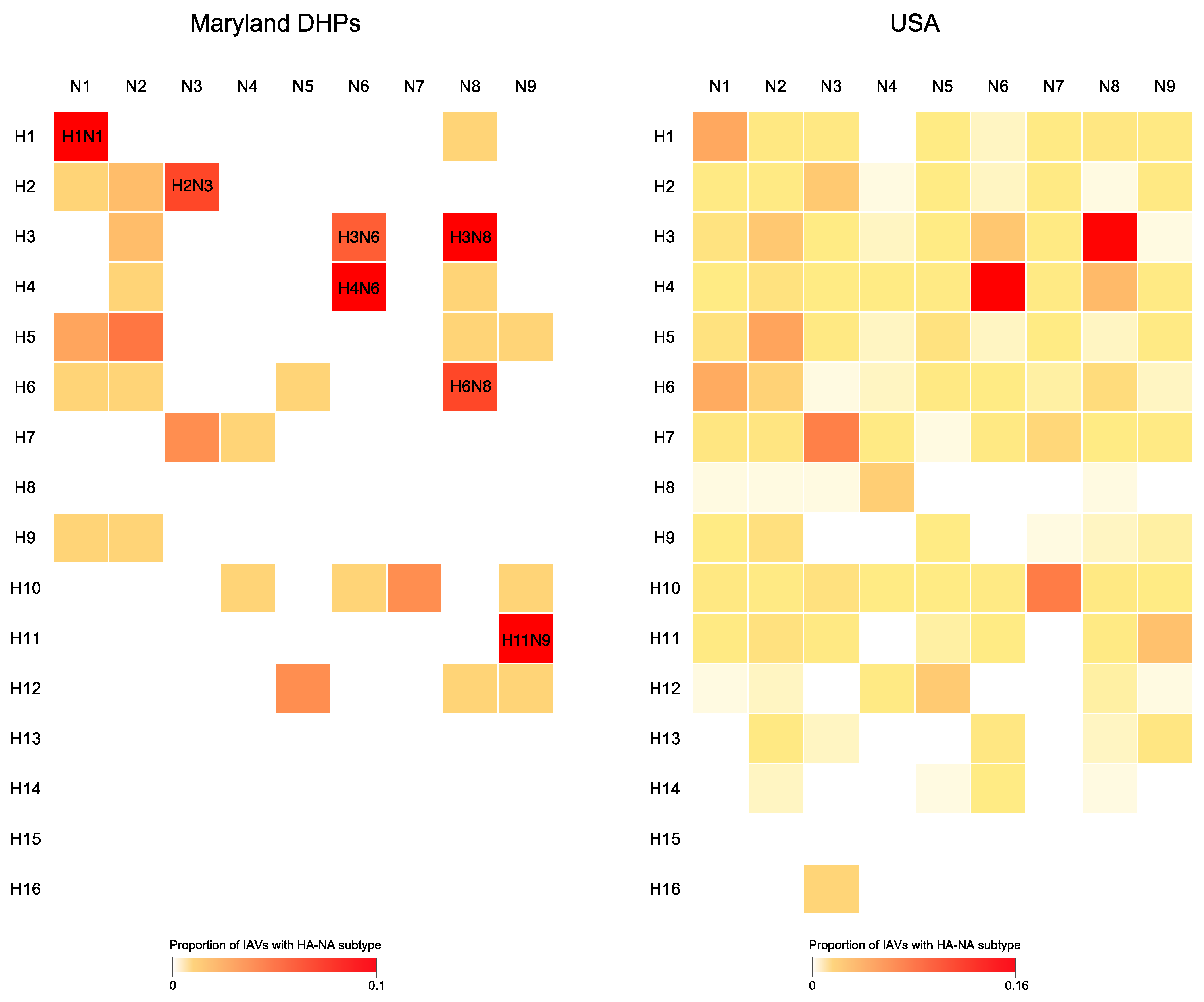
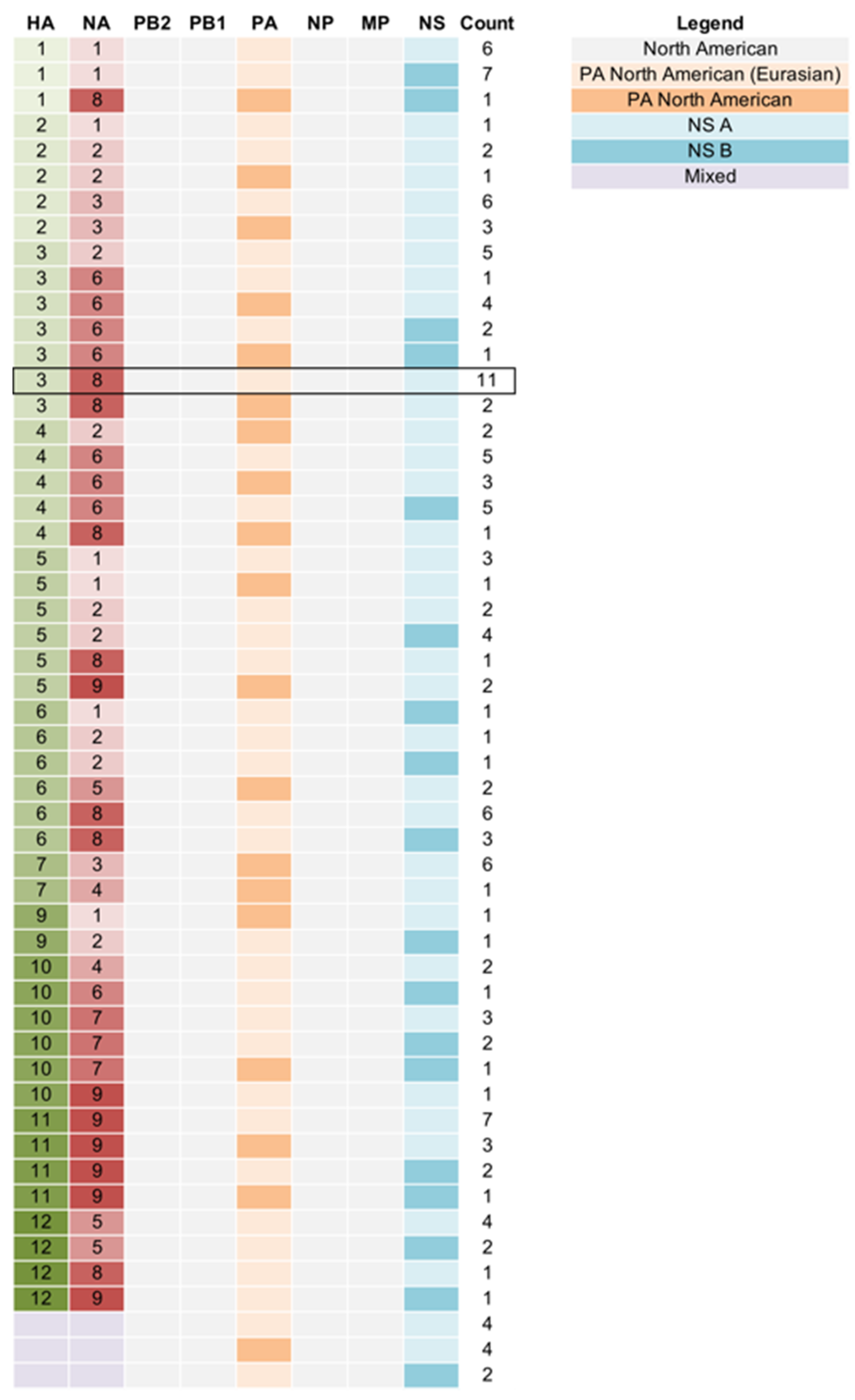
3.2. Genetic Diversity of IAV Circulating in the DHPs
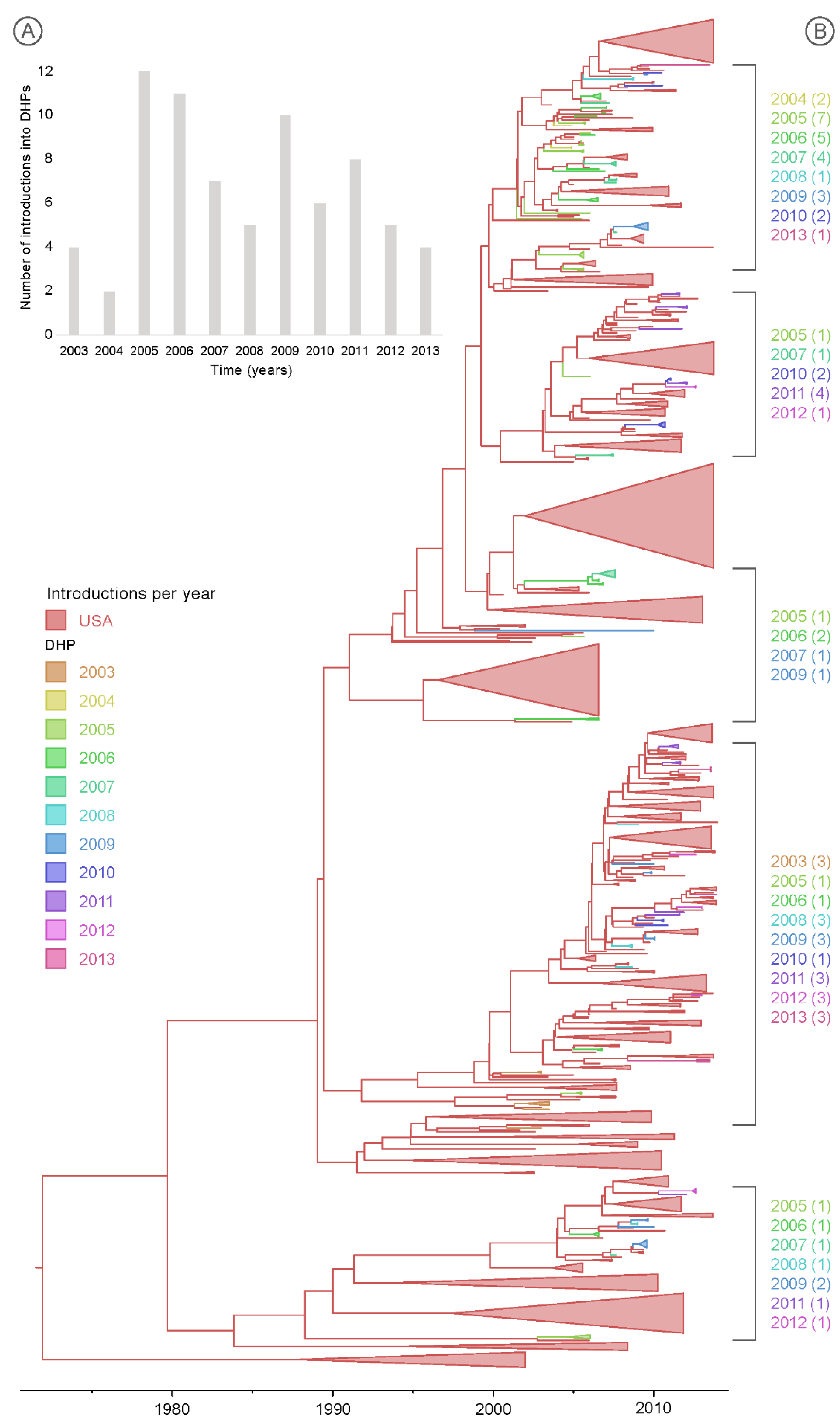
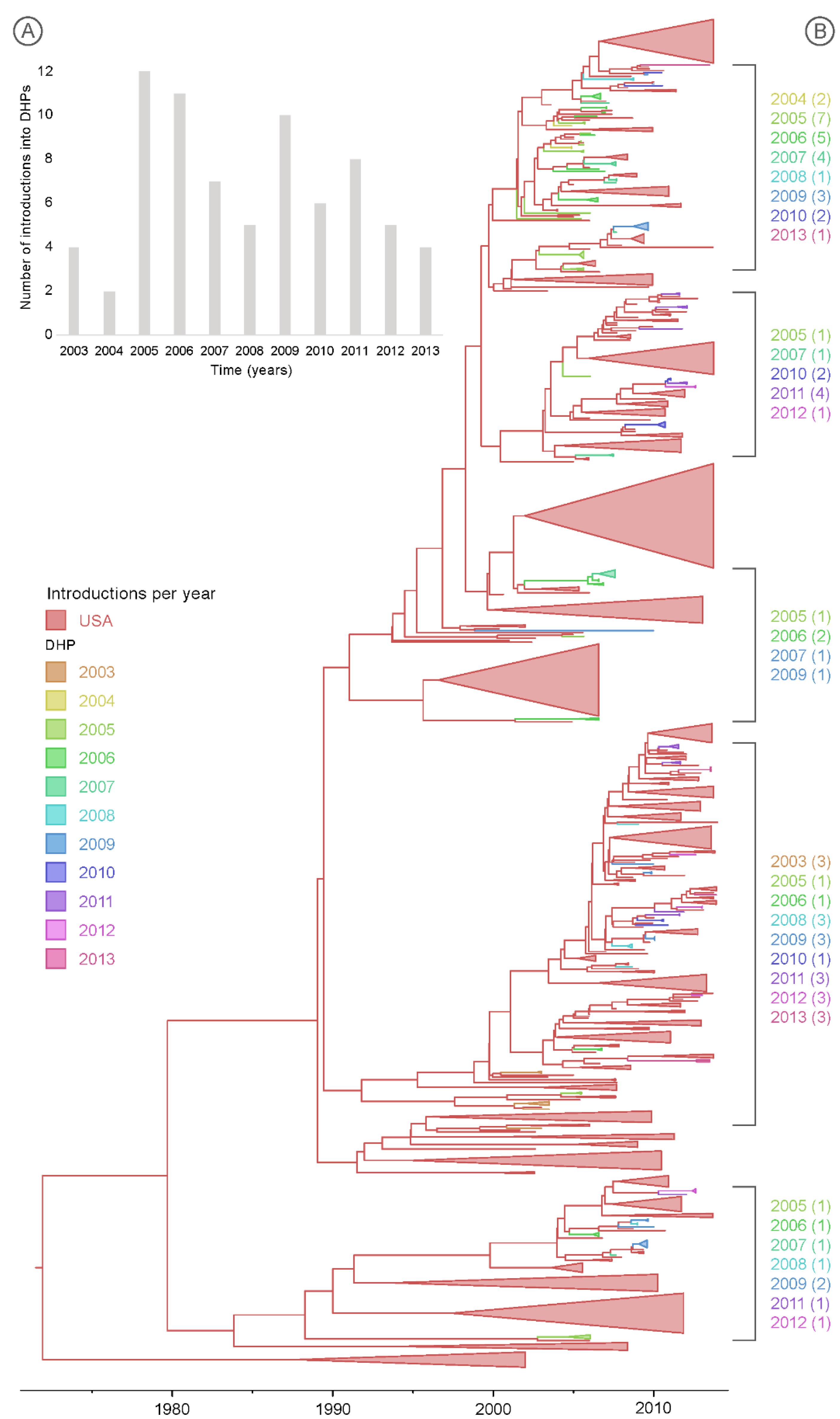
3.3. Multiple Introductions of IAV into the DHPs during the Study Period
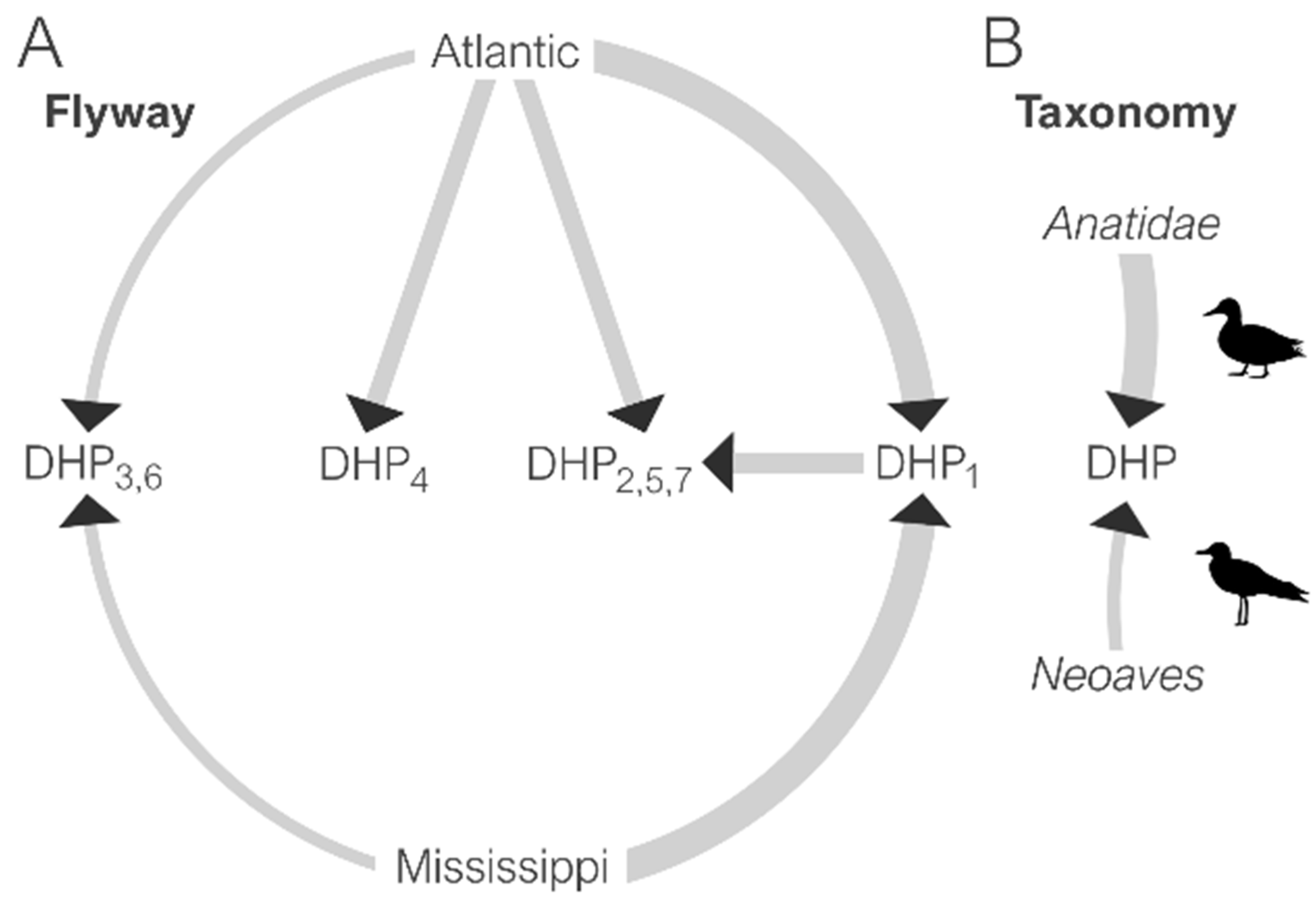
3.4. Spatial Dynamics of IAV in DHPs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Global Patterns of Influenza A Virus in Wild Birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Runstadler, J. Connecting the study of wild influenza with the potential for pandemic disease. Infect. Genet. Evol. 2013, 17C, 162–187. [Google Scholar] [CrossRef] [Green Version]
- Trovão, N.S.; Suchard, M.A.; Baele, G.; Gilbert, M.; Lemey, P. Bayesian Inference Reveals Host-Specific Contributions to the Epidemic Expansion of Influenza A H5N1. Mol. Biol. Evol. 2015, 32, 3264–3275. [Google Scholar] [CrossRef] [Green Version]
- Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Bahl, J.; Krauss, S.; Kühnert, D.; Fourment, M.; Raven, G.; Pryor, S.P.; Niles, L.J.; Danner, A.; Walker, D.; Mendenhall, I.H.; et al. Influenza A Virus Migration and Persistence in North American Wild Birds. PLoS Pathog. 2013, 9, e1003570. [Google Scholar] [CrossRef] [Green Version]
- Belkhiria, J.; Alkhamis, M.A.; Martínez-López, B. Application of Species Distribution Modeling for Avian Influenza surveillance in the United States considering the North America Migratory Flyways. Sci. Rep. 2016, 6, 33161. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.T.; Ip, H.S.; Ghedin, E.; Wentworth, D.E.; Halpin, R.A.; Stockwell, T.B.; Spiro, D.J.; Dusek, R.J.; Bortner, J.B.; Hoskins, J.; et al. Migratory flyway and geographical distance are barriers to the gene flow of influenza virus among North American birds. Ecol. Lett. 2011, 15, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Fourment, M.; Darling, A.E.; Holmes, E.C. The impact of migratory flyways on the spread of avian influenza virus in North America. BMC Evol. Biol. 2017, 17, 118. [Google Scholar] [CrossRef] [Green Version]
- Fries, A.C.; Nolting, J.M.; Bowman, A.S.; Lin, X.; Halpin, R.A.; Wester, E.; Fedorova, N.; Stockwell, T.B.; Das, S.R.; Dugan, V.G.; et al. Spread and Persistence of Influenza A Viruses in Waterfowl Hosts in the North American Mississippi Migratory Flyway. J. Virol. 2015, 89, 5371–5381. [Google Scholar] [CrossRef] [Green Version]
- Poulson, R.; Luttrell, P.M.; Slusher, M.J.; Wilcox, B.R.; Niles, L.J.; Dey, A.D.; Berghaus, R.D.; Krauss, S.; Webster, R.G.; Stallknecht, D.E. Influenza A virus: Sampling of the unique shorebird habitat at Delaware Bay, USA. R. Soc. Open Sci. 2017, 4, 171420. [Google Scholar] [CrossRef] [Green Version]
- Slemons, R.D.; Hansen, W.R.; Converse, K.A.; Senne, D.A. Type A Influenza Virus Surveillance in Free-Flying, Nonmigratory Ducks Residing on the Eastern Shore of Maryland. Avian Dis. 2003, 47, 1107–1110. [Google Scholar] [CrossRef]
- Dugan, V.G.; Dunham, E.J.; Jin, G.; Sheng, Z.-M.; Kaser, E.; Nolting, J.M.; Alexander, H.L.; Slemons, R.D.; Taubenberger, J.K. Phylogenetic analysis of low pathogenicity H5N1 and H7N3 influenza A virus isolates recovered from sentinel, free flying, wild mallards at one study site during 2006. Virology 2011, 417, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Briefings Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Biol. Evol. 2012, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Rambaut, A. Many-core algorithms for statistical phylogenetics. Bioinformatics 2009, 25, 1370–1376. [Google Scholar] [CrossRef] [Green Version]
- Pagel, M.; Meade, A.; Barker, D. Bayesian Estimation of Ancestral Character States on Phylogenies. Syst. Biol. 2004, 53, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, C.J.; Suchard, M.A.; Lemey, P.; Welch, J.J.; Barnes, I.; Fulton, T.L.; Barnett, R.; O’Connell, T.C.; Coxon, P.; Monaghan, N.; et al. Ancient Hybridization and an Irish Origin for the Modern Polar Bear Matriline. Curr. Biol. 2011, 21, 1251–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.N.; Suchard, M.A. Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 2007, 56, 391–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: http://www.R-project.org (accessed on 29 February 2020).
- Sharp, G.B.; Kawaoka, Y.; Wright, S.M.; Turner, B.; Hinshaw, V.; Webster, R.G. Wild ducks are the reservoir for only a limited number of influenza A subtypes. Epidemiol. Infect. 1993, 110, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza Virus Reassortment Occurs with High Frequency in the Absence of Segment Mismatch. PLoS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef] [Green Version]
- Dusek, R.J.; Hallgrimsson, G.T.; Ip, H.S.; Jónsson, J.E.; Sreevatsan, S.; Nashold, S.W.; TeSlaa, J.L.; Enomoto, S.; Halpin, R.A.; Lin, X.; et al. North Atlantic Migratory Bird Flyways Provide Routes for Intercontinental Movement of Avian Influenza Viruses. PLoS ONE 2014, 9, e92075. [Google Scholar] [CrossRef] [Green Version]
- Zu Dohna, H.; Li, J.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Invasions by Eurasian avian influenza virus H6 genes and replacement of the virus‘ North American clade. Emerg. Infect. Dis. 2009, 15, 1040–1055. [Google Scholar] [CrossRef]
- Ramey, A.M.; Reeves, A.B.; Donnelly, T.; Poulson, R.L.; Stallknecht, D.E. Introduction of Eurasian-Origin Influenza A(H8N4) Virus into North America by Migratory Birds. Emerg. Infect. Dis. 2018, 24, 1950–1953. [Google Scholar] [CrossRef]
- Bahl, J.; Vijaykrishna, D.; Holmes, E.C.; Smith, G.J.D.; Guan, Y. Gene flow and competitive exclusion of avian influenza A virus in natural reservoir hosts. Virology 2009, 390, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.S.; TeSlaa, J.L.; Nashold, S.W.; Halpin, R.A.; Stockwell, T.B.; Wentworth, D.E.; Dugan, V.; Ip, H.S. Evolution of a reassortant North American gull influenza virus lineage: Drift, shift and stability. Virol. J. 2013, 10, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallknecht, D.E.; Kearney, M.T.; Shane, S.M.; Zwank, P.J. Effects of pH, Temperature, and Salinity on Persistence of Avian Influenza Viruses in Water. Avian Dis. 1990, 34, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, J.H.; van Dijk, J.G.; Vuong, O.; Bestebroer, T.; Lexmond, P.; Klaassen, M. Migratory birds reinforce local circulation of avian influenza viruses. PLoS ONE 2014, 9, e112366. [Google Scholar] [CrossRef] [Green Version]
- Hill, N.J.; Ma, E.J.; Meixell, B.W.; Lindberg, M.S.; Boyce, W.M.; Runstadler, J.A. Transmission of influenza reflects seasonality of wild birds across the annual cycle. Ecol. Lett. 2016, 19, 915–925. [Google Scholar] [CrossRef]
- Stallknecht, D.E.; Luttrell, M.P.; Poulson, R.; Goekjian, V.; Niles, L.; Dey, A.; Krauss, S.; Webster, R.G. Detection of avian influenza viruses from shorebirds: Evaluation of surveillance and testing approaches. J. Wildl. Dis. 2012, 48, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Holmes, E.C. Frequent inter-species transmission and geographic subdivision in avian influenza viruses from wild birds. Virology 2009, 383, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Vittecoq, M.; Grandhomme, V.; Champagnon, J.; Guillemain, M.; Crescenzo-Chaigne, B.; Renaud, F.; Thomas, F.; Gauthier-Clerc, M.; Van Der Werf, S. High Influenza A Virus Infection Rates in Mallards Bred for Hunting in the Camargue, South of France. PLoS ONE 2012, 7, e43974. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DHP1 | DHP2 | DHP3 | DHP4 | DHP5 | DHP6 | DHP7 | |
|---|---|---|---|---|---|---|---|
| 2003 | 33/553 (6.0) | - | 102/196 (78.6) | 0/90 (0) | - | - | - |
| 2004 | 32/452 (7.1) | - | - | 6/146 (4.1) | - | - | - |
| 2005 | 22/600 (3.4) | 9/148 (6.1) | - | 0/165 (0) | 15/78 (19.2) | - | - |
| 2006 | 23/711 (3.2) | 0/115 (0) | - | 0/282 (0) | 48/169 (28.4) | 2/70 (2.9) | 5/103 (4.9) |
| 2007 | 18/618 (2.9) | 4/106 (3.8) | - | 3/347 (0.9) | 26/279 (9.3) | 1/116 (0.8) | 9/54 (16.7) |
| 2008 | 18/524 (3.4) | 42/100 (42) | - | 0/258 (0) | 2/206 (1.0) | 1/71 (1.4) | 8/98 (8.2) |
| 2009 | 50/501 (10.0) | 11/100 (11.0) | - | 3/200 (1.5) | 26/250 (10.4) | 4/120 (3.3) | 1/12 (8.3) |
| 2010 | 29/660 (4.4) | 20/100 (20.0) | - | 8/255 (3.1) | 0/100 (0) | 1/75 (1.3) | - |
| 2011 | 26/646 (4.0) | 24/150 (16.0) | - | 3/396 (0.8) | 0/150 (0) | 2/114 (1.8) | - |
| 2012 | 33/572 (5.8) | 0/100 (0) | - | 7/354 (2.0) | 28/130 (21.5) | 4/118 (3.4) | - |
| 2013 | 27/302 (8.9) | 0/61 (0) | - | 0/220 (0) | 5/120 (4.2) | 2/53 (3.77) | - |
| Total | 311/6139(5.1) | 110/980(11.2) | 102/196 (78.6) | 30/2713(1.1) | 150/1482(10.1) | 17/737(2.3) | 23/267 (8.6) |
| DHP1 | DHP2 | DHP3 | DHP4 | DHP5 | DHP6 | DHP7 | |
|---|---|---|---|---|---|---|---|
| 2003 | 5 | - | 2 | - | - | - | - |
| 2004 | 1 | - | - | 2 | - | - | - |
| 2005 | 8 | 6 | - | 3 | 2 | - | - |
| 2006 | 13 | - | - | 1 | 8 | 2 | 3 |
| 2007 | 11 | 2 | - | 1 | 2 | - | 2 |
| 2008 | 2 | 2 | - | - | 2 | - | 2 |
| 2009 | 6 | 2 | - | 2 | 5 | 3 | - |
| 2010 | 5 | 4 | - | 4 | - | 1 | - |
| 2011 | 7 | 2 | - | - | - | 2 | - |
| 2012 | 4 | - | - | 2 | 2 | 3 | - |
| 2003 | 8 | - | - | - | - | 2 | - |
| Total | 70 | 18 | 2 | 15 | 21 | 13 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trovão, N.S.; Nolting, J.M.; Slemons, R.D.; Nelson, M.I.; Bowman, A.S. The Evolutionary Dynamics of Influenza A Viruses Circulating in Mallards in Duck Hunting Preserves in Maryland, USA. Microorganisms 2021, 9, 40. https://doi.org/10.3390/microorganisms9010040
Trovão NS, Nolting JM, Slemons RD, Nelson MI, Bowman AS. The Evolutionary Dynamics of Influenza A Viruses Circulating in Mallards in Duck Hunting Preserves in Maryland, USA. Microorganisms. 2021; 9(1):40. https://doi.org/10.3390/microorganisms9010040
Chicago/Turabian StyleTrovão, Nídia S., Jacqueline M. Nolting, Richard D. Slemons, Martha I. Nelson, and Andrew S. Bowman. 2021. "The Evolutionary Dynamics of Influenza A Viruses Circulating in Mallards in Duck Hunting Preserves in Maryland, USA" Microorganisms 9, no. 1: 40. https://doi.org/10.3390/microorganisms9010040
APA StyleTrovão, N. S., Nolting, J. M., Slemons, R. D., Nelson, M. I., & Bowman, A. S. (2021). The Evolutionary Dynamics of Influenza A Viruses Circulating in Mallards in Duck Hunting Preserves in Maryland, USA. Microorganisms, 9(1), 40. https://doi.org/10.3390/microorganisms9010040