1. Introduction
Mycobacterium (M.) avium subsp.
paratuberculosis (MAP) is a globally important obligate pathogen of domestic and wild ruminants, causing a chronic progressive granulomatous enteritis designated as Paratuberculosis or Johne’s disease [
1]. First reported at the end of the 19th century in Europe, Johne’s disease spread throughout the world, particularly in bovine dairy industries, and was later also diagnosed in sheep and goats in many countries [
2]. There are strain dependent differences in disease progression in cattle and small ruminants like sheep and goats [
3]. Knowledge of genomic features that convey relative virulence and pathogenicity of MAP types in different hosts is likely to help with devising control strategies and methods.
MAP can persist in the environment and has been isolated from raw milk, different non-ruminant species and humans. The evidence of a zoonotic capacity of MAP concerning Crohn’s disease in humans is under ongoing discussion [
4]. Together with the obligate bird pathogens
M. avium subsp.
avium (MAA) and subsp.
silvaticum, the facultative-pathogenic and genetically variable pool of organisms known as
M. avium subsp.
hominissuis (MAH), and the feline pathogen
M. avium subsp.
lepraemurium, MAP belongs to the species
M. avium and to the
M. avium complex (MAC) [
5,
6,
7]. Strains of MAH cause disseminated infections, pulmonary diseases and lymphadenitis in humans, and granulomatous lesions in pigs and many other animal species with wide-ranging environmental sources of infection [
8,
9,
10,
11,
12,
13]. The majority of human infections occur in immunocompromised people, persons with underlying pulmonary diseases and children with cystic fibrosis [
14]. In recent years, MAH represents an increasing public health concern in developed countries [
15].
Based on genotyping and on phenotypic features including different host associations and growth characteristics, MAP strains have been divided into two main groups: The Cattle-type (MAP-C) also designated as type II which also includes Type B, and the Sheep-type (MAP-S), which is further subdivided into sub-groups type I, III and sub-lineages of camelid isolates [
16,
17]. These classifications were recently confirmed by whole genome sequence analyses (WGS) based comparisons of single nucleotide polymorphisms (SNPs) [
18] in draft genomes of numerous MAP strains from diverse locations worldwide.
When trying to use genomics to understand evolution and why some strains are more virulent/pathogenic than others in different hosts, ideally multiple, completely closed genomes of each important type from diverse regions should be compared. However, whole genome sequencing and assembling of MAP is hampered by some major technical challenges. Depending on differences in media, the number of passages and the genotypes of individual strains, cultivation of these bacteria for WGS requires 4–12 weeks (MAP-C) or 4 weeks to 7 months (MAP-S) [
19,
20]. MAP genomes have an extremely high GC content (close to 70%) that causes biases in PCR amplification steps [
21,
22,
23] during the preparation of Illumina libraries creating uneven read representation within the genome sequences [
24,
25,
26]. The assembly of such regions is often inefficient resulting in gaps, if libraries are prepared following standard protocols [
27]. This is further complicated by the presence of numerous repeated sequences, which make assembly even more challenging.
Currently, twelve complete MAP-C genome sequences, one human and eleven bovine-derived, from four different regions of the world, have been published. These include MAP K-10 [
28], MAP4 [
29], and FDAARGOS_305, which originated from U.S., MAP E1 and E93, which originated from Egypt [
30], MAP/TANUVAS/TN/India08 (GenBank accession no. CP015495.1), five MAP genome sequences from South Korea (GenBank accession no. CP033909-10, CP033427-28), and MAP JII-1961 from Germany [
31]. Recently, a MAP-S/type I strain (Telford) from Australia was completely closed by a combination of Illumina and PacBio sequence technology [
32]. Although, there are MAP-S/type III draft genomes available—for example strain S397 with 176 contigs from U.S. [
33], and strain JIII-386 with six scaffolds from Germany [
34] as well as additional incomplete MAP genome sequences published—to date there are no completely closed MAP-S/type III genomes.
Results of a very recently published comprehensive pan genome and phylogenetic analysis of 28 MAP and non-MAP genomes (including MAH and MAA strains) support the prediction that the core genome evolves through SNPs and recombination events while the accessory genome is acquired by horizontal gene transfer [
35]. In contrast to the non-MAP strains, the included MAP-C genomes were very stable with a low number of SNPs and accessory genes, and a lack of rearrangements [
35].
Due to limitations with early versions of genomic analysis software, deciphering MAC evolution based on the presence, absence and order of genes has proven to be problematic. Major genomic differences identified as insertions or deletions among strains of MAC have been designated as large sequence polymorphisms (LSPs) [
36,
37]). Based on the presence of specific insertions, deletions and inversion events of LSPs, the evolutionary relationship among MAC strains was determined and a biphasic or triphasic evolution of modern MAP type strains was proposed [
33,
38,
39]. However, six genomic insertions were found only in MAP and not in the other MAC members (LSP
P 2, 4, 11, 14, 15, and 16). In addition, genes of a 10 kb LSP (LSP
A8) were absent from all MAP lineages [
38]. A loss of three regions (LSP
A4-II, MAV14, and LSP
A18) was found to be characteristic of the MAP cattle lineage, and two deletions (LSP
A20 (deletion #1), and deletion #2) were specific for the MAP sheep lineage [
34,
38]. Based on all these analyses and specific in silico observed inversions, sheep type isolates seem to be an intermediate in the evolution of MAP-C type strains by the emergence from a pathogenic clone (proto-MAP) which was closely related to MAH or
Mycobacterium intracellulare strains [
33]. Another study [
34] challenged the suggestion that
M. intracellulare was an ancestor of proto-MAP and explored how well new genomic evidence fit with the previous alternative hypotheses suggesting either evolutionary division of proto-MAP into MAP-S and MAP-C or a successive evolution from such a clone via MAP-S to MAP-C. Currently there is no clear evidence that an ancient MAH subspecies was a progenitor of MAP [
35].
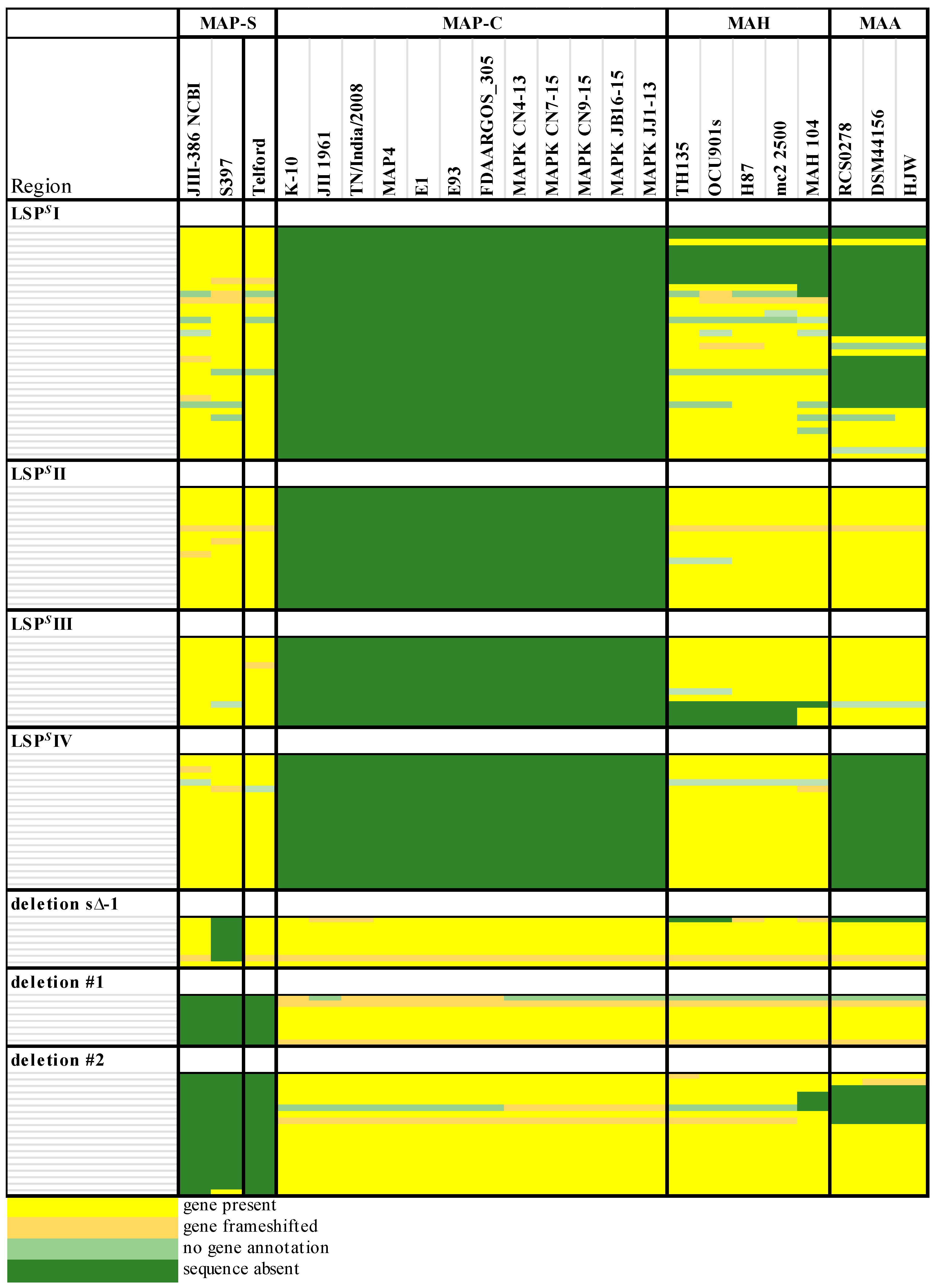
Genomic differences between MAP-C and MAP-S-type isolates are also evident but not fully defined due to the lack of finished MAP-S genome sequences of isolates from different origins. Based on three sheep strain draft genomes (MAP-S/type III) from the U.S., Bannantine et al. [
33] proposed ten probable MAP-S specific LSP
Ss, which could not be detected in strain K-10 (MAP-C). Using alternative analytical techniques and including an additional unfinished sheep strain from Germany, Möbius and colleagues [
34] disputed half of these suggested LSP
Ss because they found four putative MAP-S specific regions were fully and one partly detected in MAP K-10, MAP4 and JII-1961 genomes (all MAP-C). This study extended, merged and re-defined the previous MAP-S specific described regions [
38] and [
33] into: LSP
S I to LSP
S IV. In addition, genes that were apparently absent in sheep type strains from the U.S. and defined as a MAP-S specific deletion [deletion s∆−1] [
40], were later identified in MAP-S strains from Australia and Germany [
34]. Former studies discussed the existence of a third major lineage within MAP besides the cattle and the sheep type group (MAP-C and MAP-S): the type III or intermediate group, exhibiting differences regarding host association and growth characteristics [
16]. Phylogenetic analysis based on sequence comparisons of
gyrA,
gyrB, and
recF [
41], or of genome wide homologous CDS (conserved at both, the nucleotide and amino acid level) and ncRNAs [
34] revealed the subdivision of MAP-S strains into sub-groups. In addition to the previously repeatedly characterized type I, and III lineages [
39], there exists a third sub-lineage, comprising MAP genomes of isolates from Arabian camelids (JQ5 and JQ6; [
17]). As mentioned above, this MAP-S subdivision was clearly confirmed based on SNP analysis from whole genome sequences of 20 MAP-S and numerous MAP-C strains of different origins worldwide, presented in a large phylogenetic tree [
18]. This tree emphasized the higher diversity of MAP-S relative to MAP-C strains. Currently there is a knowledge gap about type I and type III specific insertions/deletions of genes and gene clusters.
For the present work, the novel Nanopore-long sequence read technology was employed to completely close the genome of German MAP-S/type III strain JIII-386 using previously obtained Illumina whole-genome shotgun sequence data to polish the Nanopore derived sequence. Nanopore is relatively inexpensive technology and is especially promising for high GC-content WGS, since no PCR amplification is required for the preparation of Nanopore sequencing libraries. Nanopore sequencing technology, developed by Oxford Nanopore, is one of the latest DNA sequencing methods and represents a third-generation approach. Using Nanopore sequencing, a single molecule of DNA is transported through a nanometer large pore [
42]. These nanopores consist of recombinant proteins embedded in a polymer membrane. A bias voltage is applied across the membrane. Nucleotides that passes through the nanopore create voltage changes that are specific for each of the four nucleotides, enabling the DNA sequence to be read out. Single DNA molecules longer than a megabase can be sequenced using Nanopore, but the resulting sequence has a rather high error rate (usually in the 5–20% range). By means of sequence assembly and polishing of the consensus sequence with high depth Illumina reads, a similar error rate in comparison to short read data can be reached [
43] but provides sequence over repeat regions and other regions that are not well defined with short read methods.
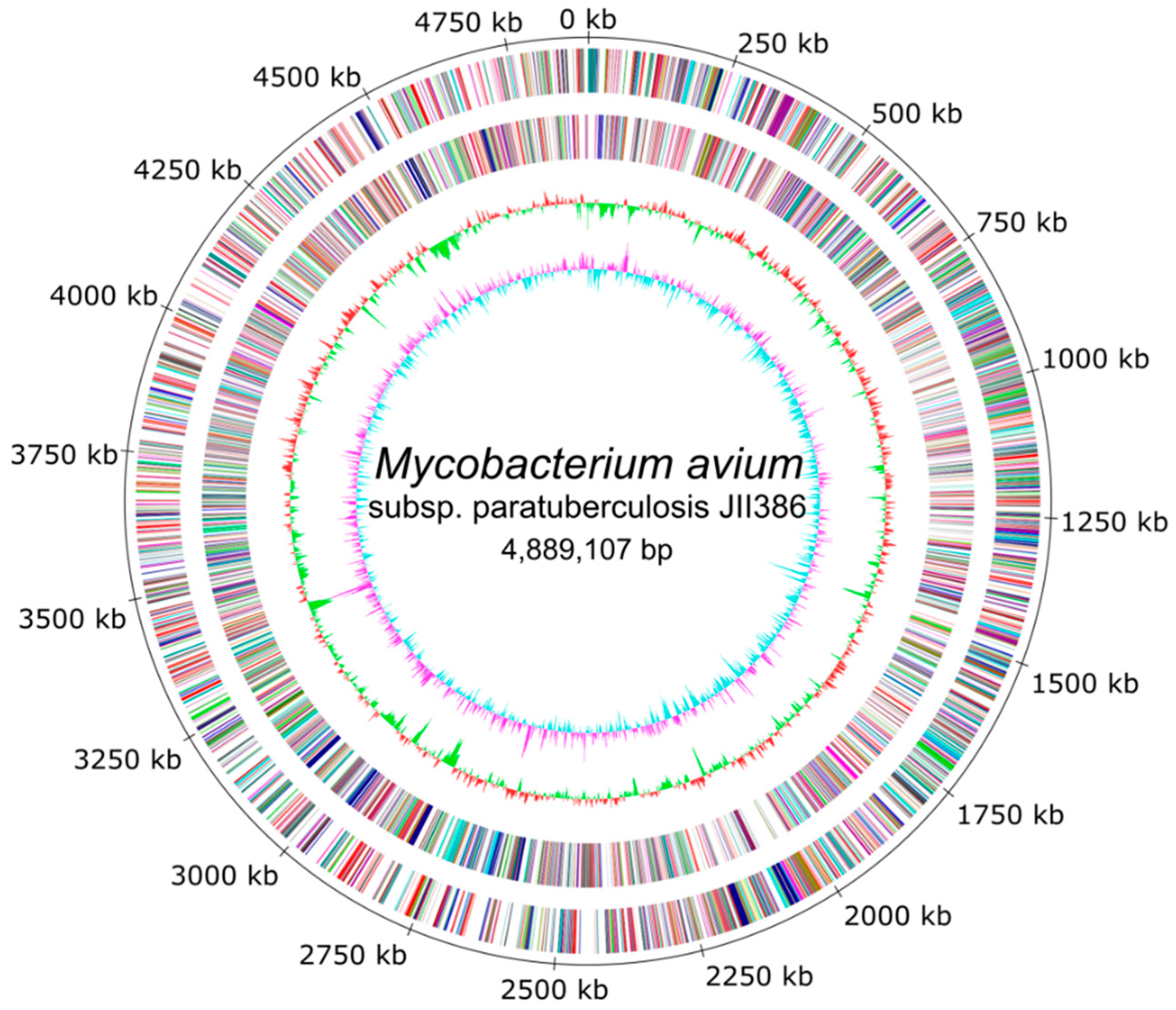
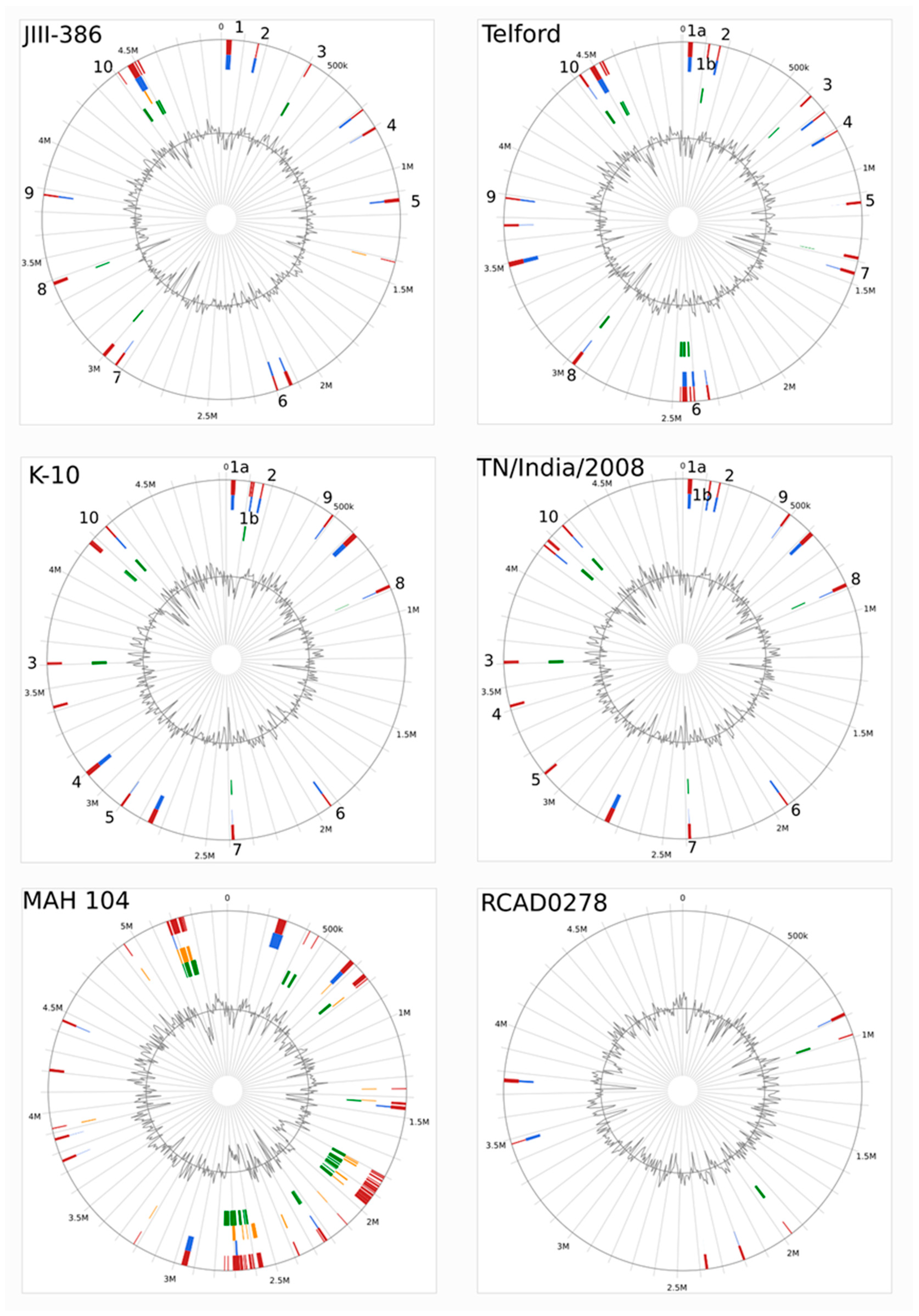
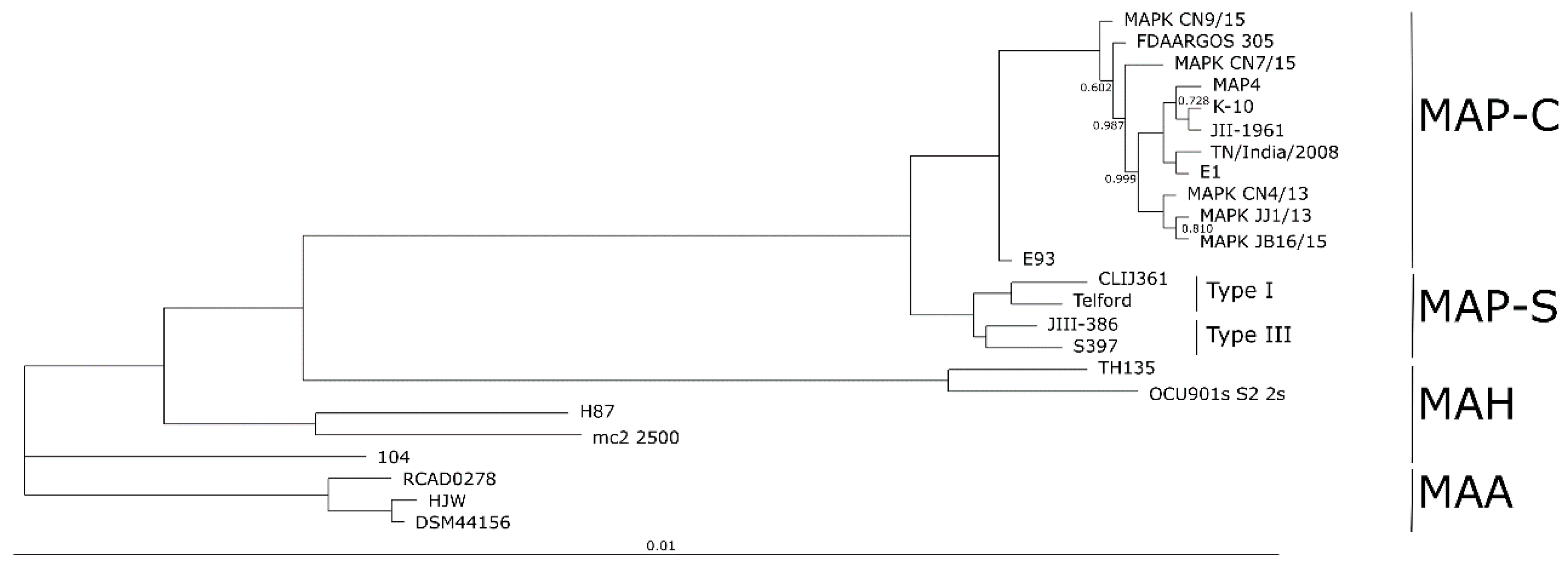
The completely closed and annotated genome sequence of JIII-386 is described here. Many genomic features of JIII-386 are compared to that of the recently published closed MAP-S/type I strain Telford, the MAP-C reference genome K-10, and the draft genome of MAP-S/type III strain S397. These searches include comparisons of the gross genomic structures, numbers of SNPs and INDELs, presence of specific genomic elements, selected putative virulence associated genes, secondary metabolite and antibiotic resistance gene clusters, and the content and localization of genomic islands. Furthermore, the phylogenetic relationship of a selection of MAC genomes including available and recently published complete MAP-C, MAA, and five closed MAH genomes of different geographic origin has been calculated and presented. MAP-S specific LSPSs and deletions are examined based on this enlarged strain panel. Comprehensive core and pan genome analyses assisted in clarifying and confirming differences identified previously to define typical and possibly new characteristics of MAP-S and MAP-C strains and especially of MAP-S/type I and type III genomes within the MAC based on new finished genome sequences.
4. Summary
A major goal of this study was to close the draft genome of the German ovine MAP strain JIII-386 using the relatively novel Nanopore Technology and polish this closed genome with deep whole-genome shotgun Illumina paired-end sequencing data. Based on this new closed MAP-S/type III strain JIII-386 and the recently published complete genome sequence of MAP-S/type I strain Telford from Australia, the present study aimed to close knowledge gaps concerning genomic differences between MAP-S subgroup I and III, and MAP-S and MAP-C (type II) strains, and to reveal new insight into MAP genomics and evolution. Although all bioinformatic methods used for the genome-wide comparisons are optimally applied to closed complete genome sequences, the high-quality draft genome of type III strain S397 from U.S. was also analyzed to increase the number of MAP-S genomes. In addition to MAP-S genomes, the study included twelve MAP-C, three MAA and five MAH genomes, all closed and annotated by NCBI PGAP.
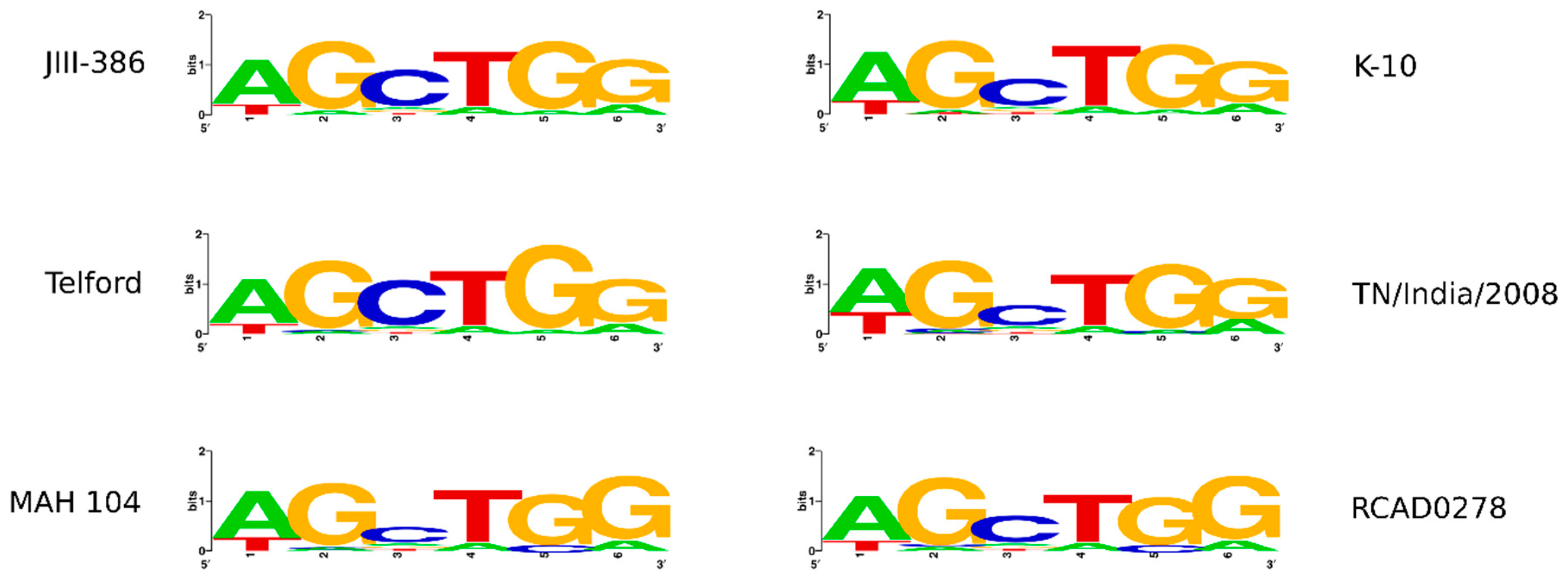
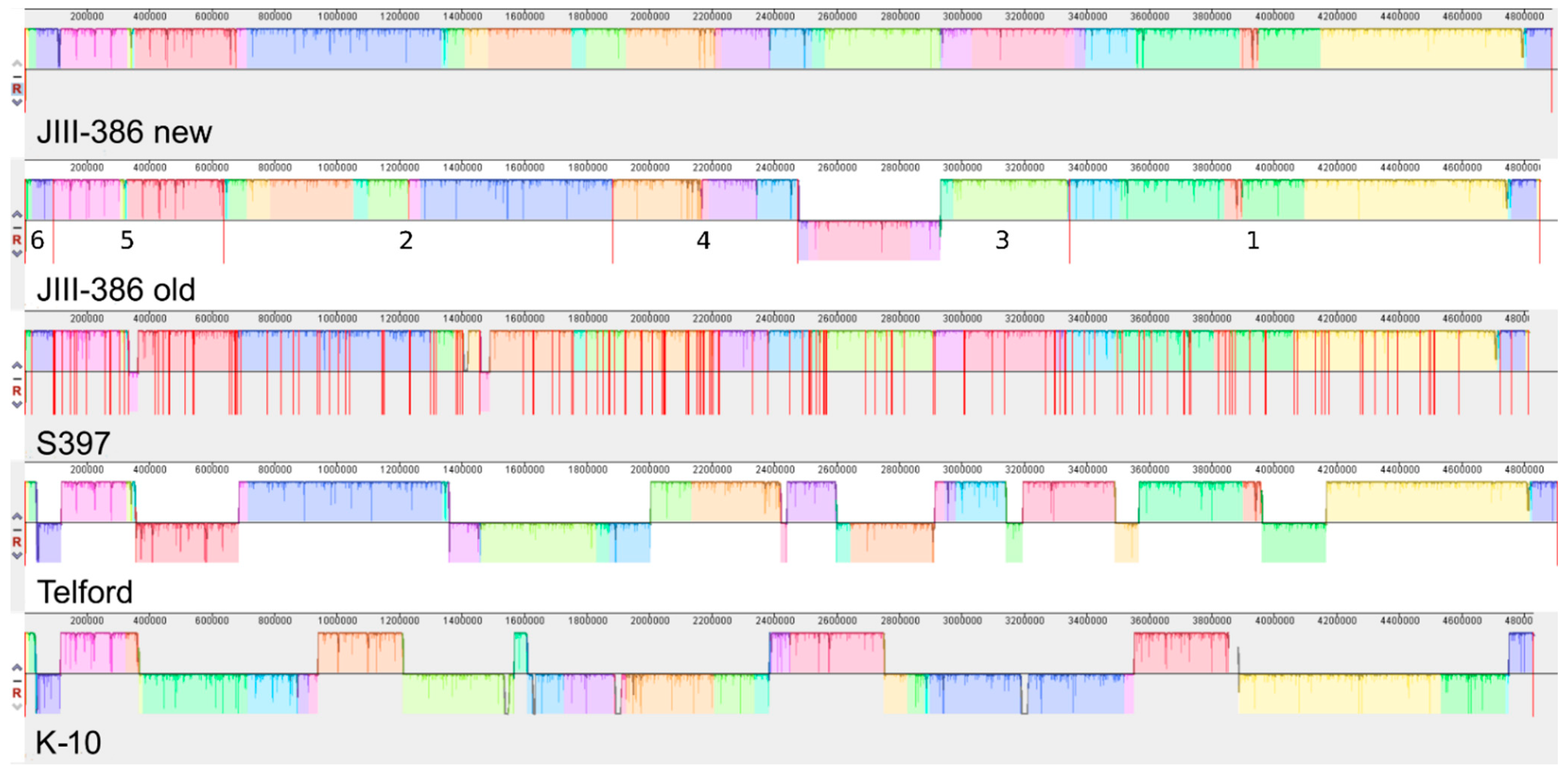
In the first part of the study, general genomic features of the newly closed JIII-386 were described and compared with the draft genome of JIII-386 and S397 and the completed genomes of Telford and K-10. MAP-S/type I strain Telford is the largest genome here comprising more CDS and genes with predicted function than MAP-S/type III and MAP-C genomes. The Shine–Dalgarno sequence motif 5′-AGCTGG-3′ suggested to be characteristic for Mycobacteria [
34], was confirmed for new closed MAP-S strains, and also for different other MAP-C and for MAA strains which may be helpful in future bioinformatic studies. The structural comparative alignments (MAUVE plots) among JIII-386, Telford and K-10 genomes and also the distribution of corresponding genomic islands clearly reveal the mosaic nature of MAP genomes with different type and subtype. Specific genetic elements such as CRISPER loci, fragments of putative prophages and like regions, IS elements, and genomic islands were identified, and in many cases confirmed by using multiple approaches to avoid misrepresentations. The comparison of JIII-386, Telford, and K-10 revealed many conformities. These different MAP-subtype genomes contained no functional CRISPR-Cas region and only one large potential fragment (17.7 kb) of a prophage genome. JIII-386, Telford and K-10 share many secondary metabolite compound clusters. None of the different MAP type genomes harbored any typical antibiotic resistance genes, but all of these strains harbored a homologous gene for a known ABC-type multidrug transport system that is involved in drug resistance in
M. tuberculosis. Comparisons of selected putative virulence genes and gene clusters (
mce,
mmpL,
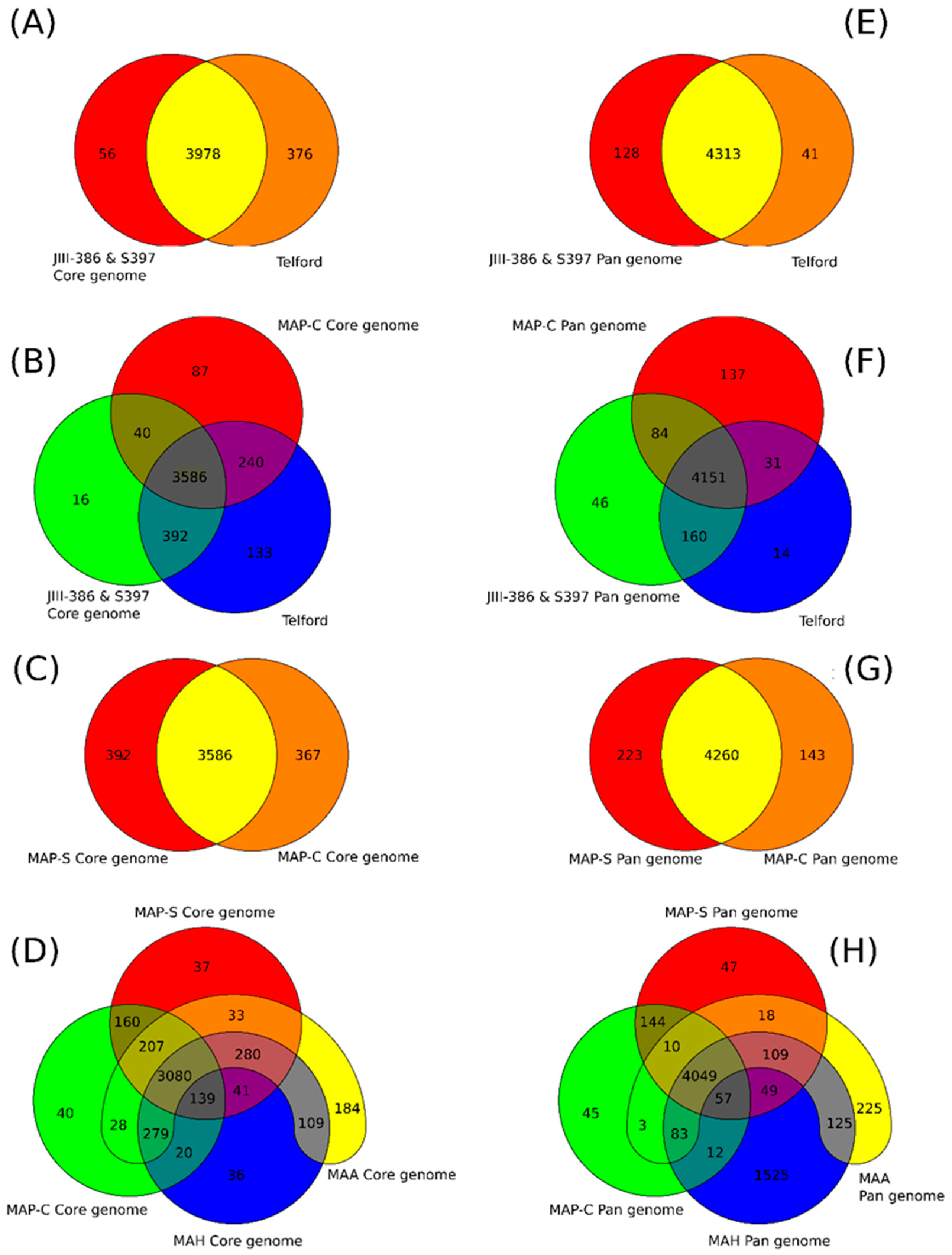
rnd, and PE/PPE genes; and Mycobactin gene cluster) show a high conformity but also differences concerning the presence, the length, the sequence and the completeness of individual genes as summarized below. Comprehensive core and pan genome analysis based on the enlarged genome panel, revealed the large number of highly conserved common genes but also unique genes (accessory genes) probably acquired by horizontal gene transfer in different MAP-types.
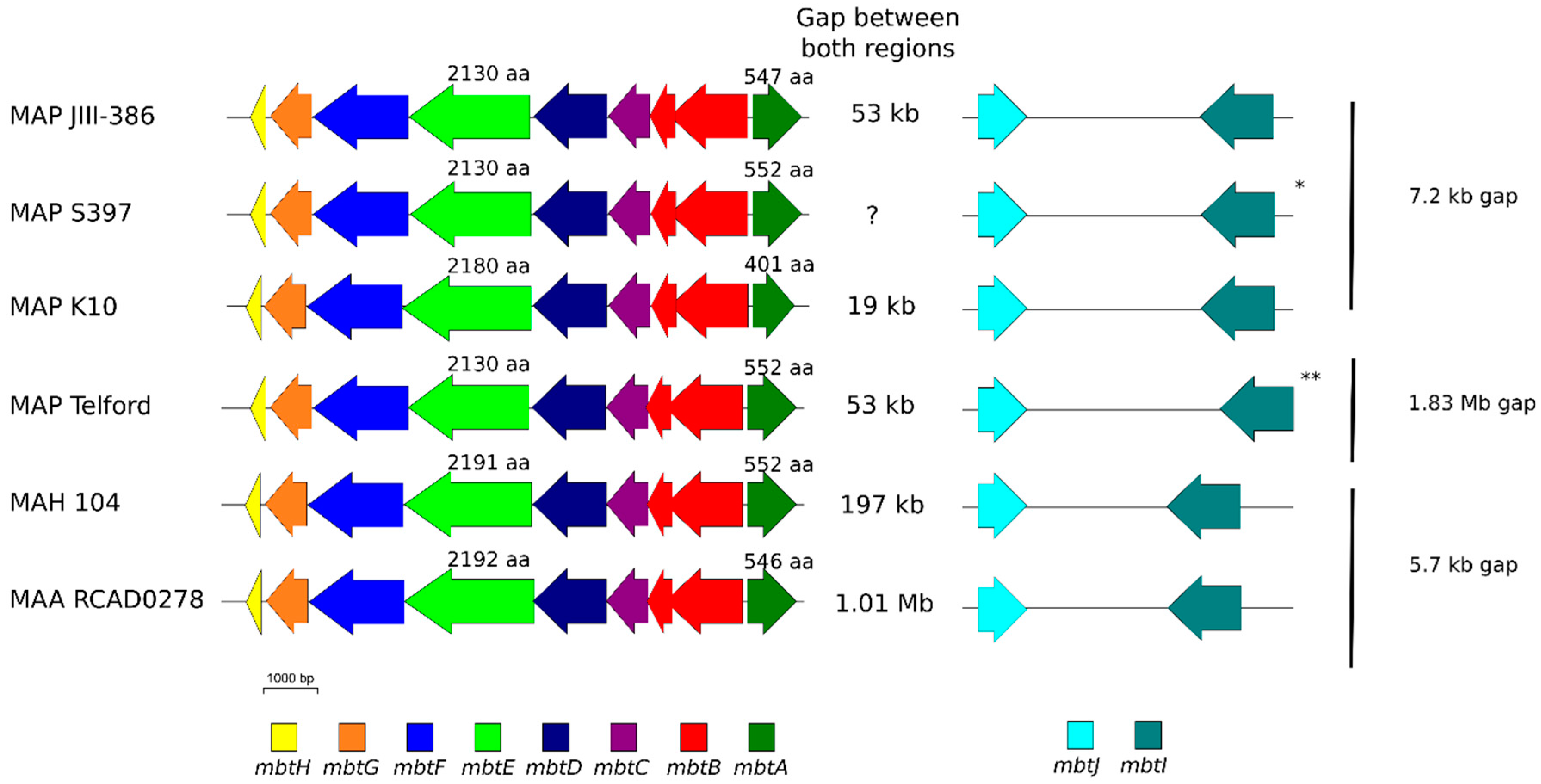
The phylogenetic tree based on core genome of 24 M. avium genomes clearly illustrates the distinct grouping of MAP-S and MAP-C as well as MAP-S type I and III sub-clusters. In addition, the SNP analyses conducted for this work support the tree results. The study confirmed the results of earlier previous reports that major genetic differences between MAP-S and MAP-C group include genes of the known large sequence polymorphisms (LSPSs) regions I–IV which are absent in MAP-C, and genes of deletions #1 and #2 which are absent in MAP-S, which could potentially account for the major type specific phenotypic features. Additional differences were uncovered. Type I and type III MAP-S genomes showed a higher structural diversity in contrast to MAP-C genomes, which exhibited a strong synteny of chromosomal structure. The number of specific IS elements (IS900, IS1311, IS3 family, and ISMap02) differed; in total more IS elements were detected in MAP-S than in MAP-C genomes. Many more hints of differences between MAP-S and MAP-C genomes were confirmed and new ones identified due to detection of known virulence-associated genes. Both MAP-S subtype strains harbored two mce CDS that were outside of detectable mce operons, whereas MAP-C genomes harbored only one. The current analysis confirmed that the MAP-C strains includes an additional mmpL gene that was absent in both MAP-S genomes. There were individual PPE genes which were lost in one or the other MAP-type. MAP-type specific differences in the Mycobactin gene cluster were determined that could possibly explain sub-type specific differences in growth characteristics. Compared to K-10 (MAP-C), in type I and III strains of MAP-S the gene mbtA is not truncated, the gap between mbtA and mbtJ is larger (53 kb vs. 19 kb), and mbtC is interrupted. Furthermore, the gene mbtE gene is shorter and as with mbtA shows only up to 40% sequence identity to the same genes in K-10. Moreover, MAP-C genomes include an additional secondary metabolite gene cluster. The core genome analyses uncovered many unique genes in MAP-S or MAP-C core genome, absent or frameshifted in the core or the pangenome of the other group. These include genes encoding cytochrome 450, different cytochrome subunits and different type VII secretion proteins, which are likely to be associated with virulence. Remarkably, in the core genome of MAP-C, two genes were identified encoding for cytochrome 450 proteins (MAP_RS00055 and MAP_RS06975), without any homolog in the MAP-S pan genome. MAP-S genomes contained two additional small gene clusters of 2 kb each with three genes, that were absent in MAP-C genomes possibly lost during MAP-C evolution.
Another main objective was to clarify which genetic differences assist with subdivision of MAP-S into type I and type III subgroups. In summary, the following results support the subdivision: (1). Structural comparative alignments (MAUVE plots) show a relatively strong synteny among both MAP-S/type III genomes (JIII-386 and S397) but much more rearrangements and inversions between type III and Telford (type I) genomes. (2). The SNP analysis revealed a lower number of SNPs in comparison of the two type III genomes than in a comparison of type III and type I genomes. (3). The genome of type I strain Telford is larger; contains more IS elements and a higher number of GIs were identified in Telford than in type III strain JIII-386 suggesting different evolutionary adaptation processes. (4). Within the mycobactin cluster, JIII-386 and S397 showed different sizes for the gap between mbtJ and mbtI when compared with Telford (5). The search for unique genes in type III strains (JIII-386 and S397) and or Telford, revealed 37 and 27 genes respectively, and these were predicted to encode several virulence associated proteins. All results of these comparisons will need to be re-examined when more closed MAP-S type genomes become available.
Because the divergence of MAP subtypes from their common ancestor is a relatively recent event in the evolution of M. avium, there are fewer genomic differences relative to other MAC bacteria. Results from the current and previous studies hint at the evolutionary origin of MAP-S/type I, type III, and MAP-C/type II subgroups in relation to MAH and MAA. Results of the current analyses confirmed that MAP-S strains are more diverse than MAP-C strains. In addition, the larger genome size of Telford, and the higher number of IS elements and GIs, compared to JIII-386, suggest a possible earlier emergence of type I than type III. The phylogenetic tree presented here suggested a slightly earlier emergence of MAP-S strains from a proposed common progenitor than MAP-C strains. MAP-C strains seem to have emerged more recently, possibly because of intensive cattle farming combined with an increasing MAP contamination of the environment as well as distribution across the world by intensive livestock trade. MAA and MAP are pathogens that are well adapted to their hosts, but MAA is more closely related to MAH than to MAP. The understanding of the emergence of obligate human and animal pathogens possibly from more ubiquitous occurring, facultative pathogenic members of the MAH group is very important in a world which is burdened by overpopulation and profound changes to the environment including those occurring in pathogenic and non-pathogenic bacteria.
In conclusion, here we compared genome structure, virulence associated genes, conserved genomic island and core genomes of available complete or nearly complete MAP genomes and close MAC relatives. We found that the few available MAP type S genomes are distinct from MAP C types. The genomic variations among MAP-S type III strains collected from different geographic locations were small enough to form a close cluster and the genomic variations among the strains in MAP-S types I and type III are big enough to form two distinct groups. Major limitations of these analyses are the paucity of differences due to the very slow tendency of MAP to acquire genomic changes relative to other bacteria and a lack of representative S type references. These groupings will become more robust as more type I and type III S genomes from different parts of the world become available for use as references.
However, the finished S type genome sequences of JIII-386 (type III) and Telford (type I) that were scrutinized here provide a valuable resource for further M. avium research. The comparison of MAC genomes from the current study provide a reference of MAP specific similarities and differences that may help to improve our understanding of mycobacterial pathogenicity and evolution.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}