On-Site Blackwater Treatment Fosters Microbial Groups and Functions to Efficiently and Robustly Recover Carbon and Nutrients

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
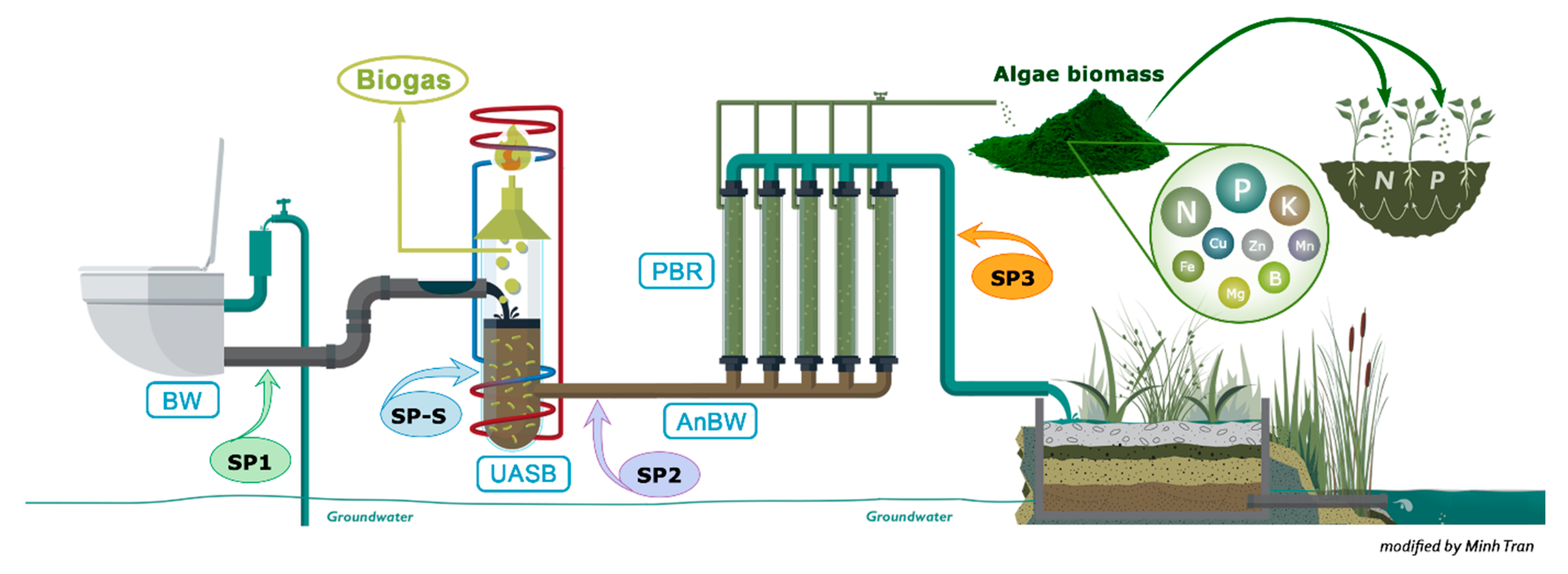
2.1. Blackwater Samples
2.2. Chemical Analysis
2.3. DNA Extraction
2.4. DNA Shotgun Metagenome Analyses
2.5. Statistical Analysis
3. Results
3.1. Physicochemical Composition of the Blackwater Anaerobic Digestion System
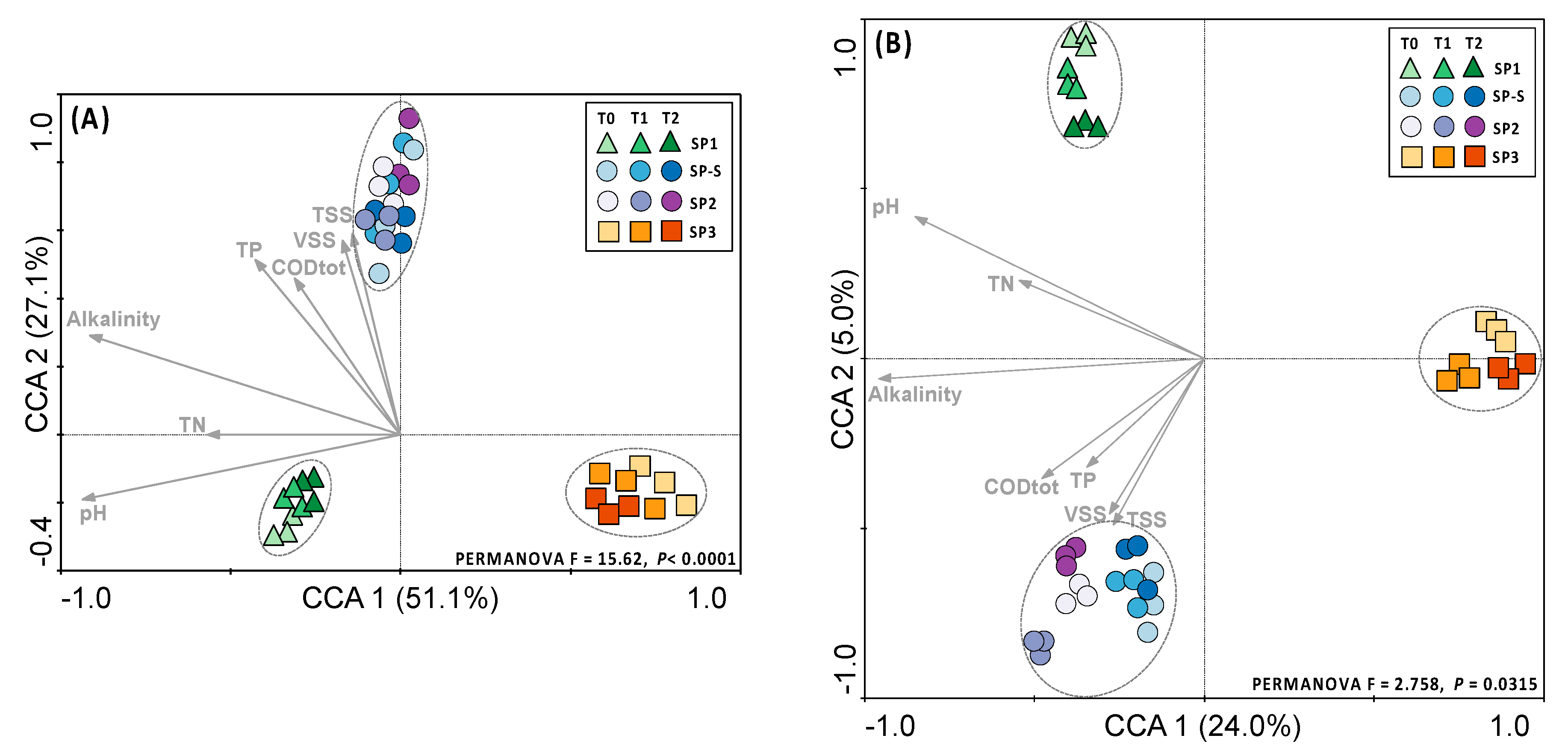
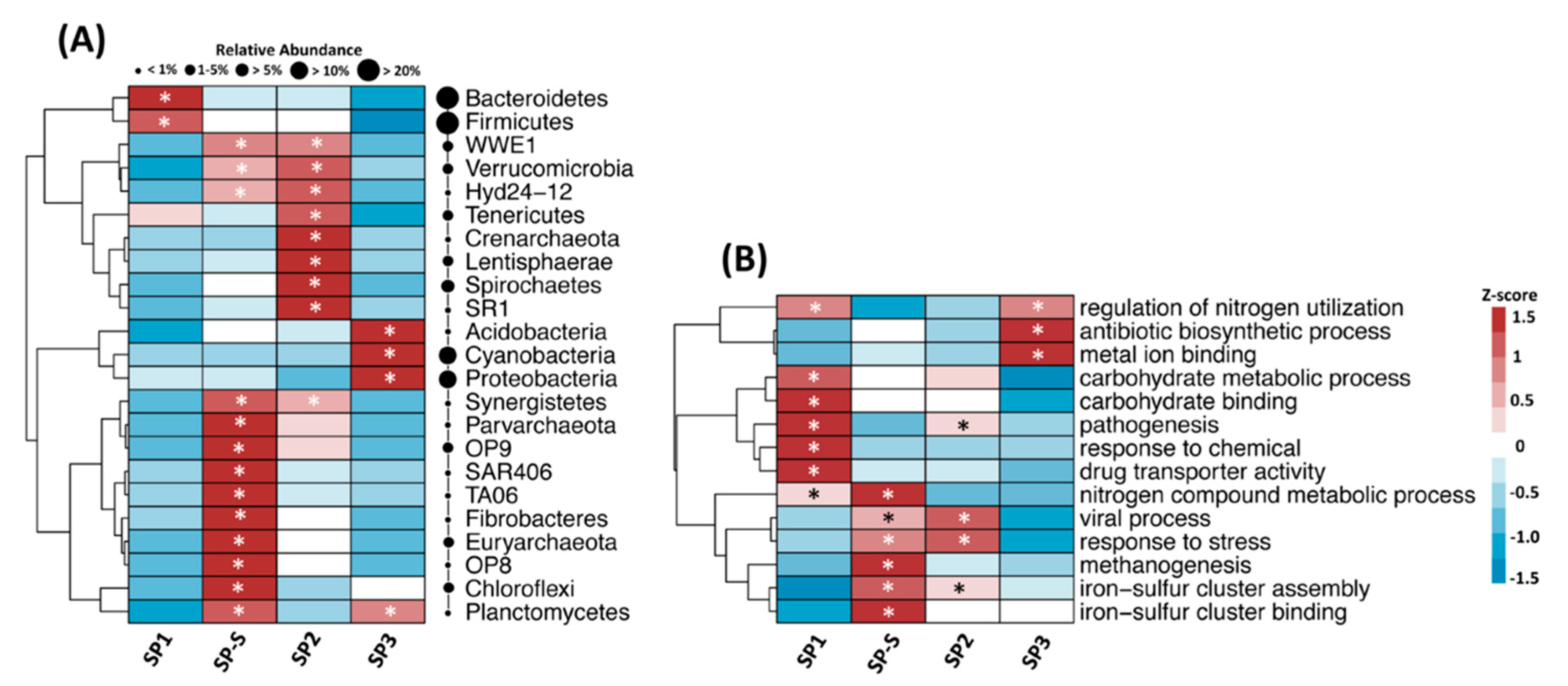
3.2. Microbial Community Structure
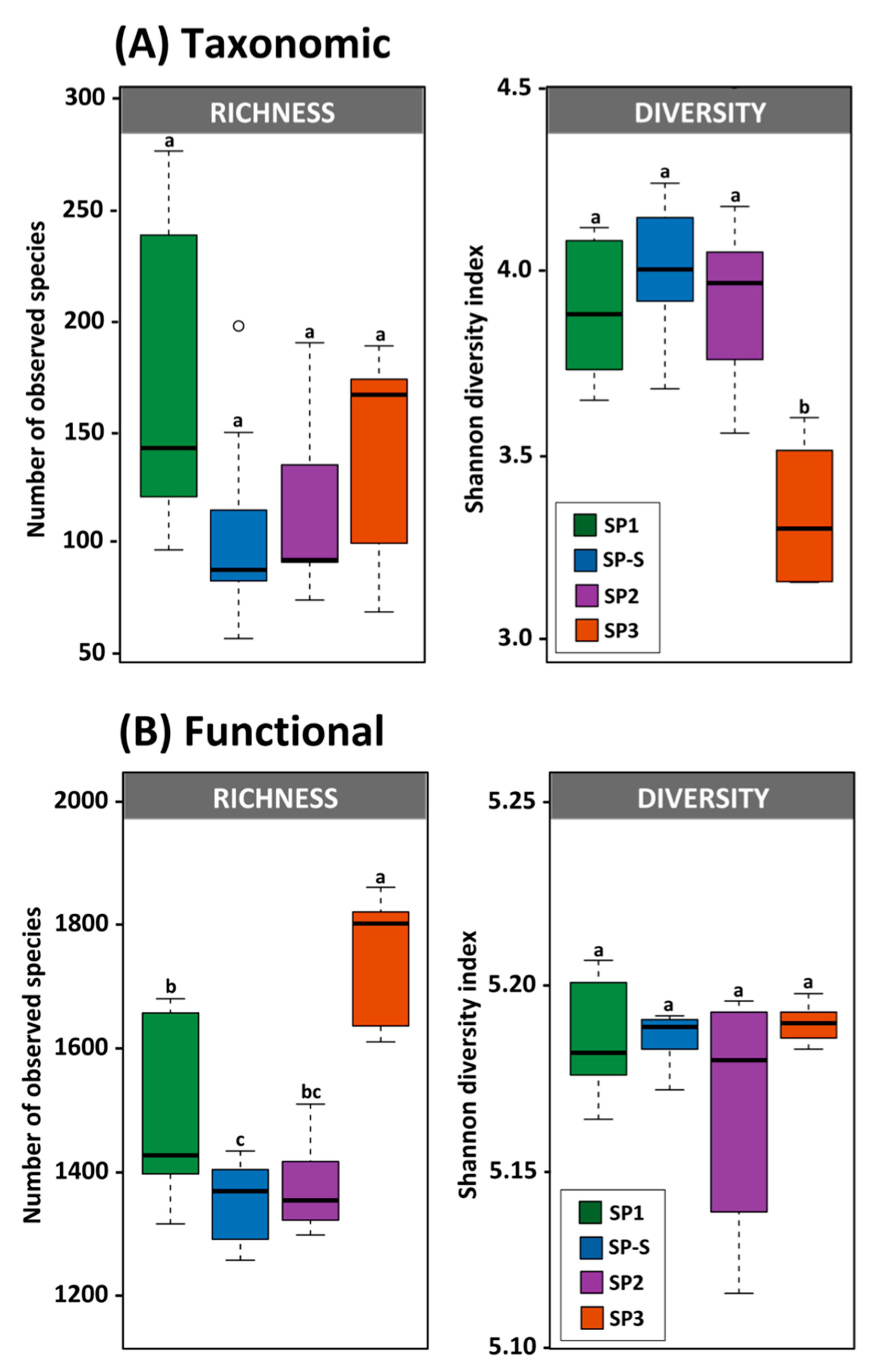
3.3. Microbial Community Diversity and Composition
3.4. Microbial Functional Profile
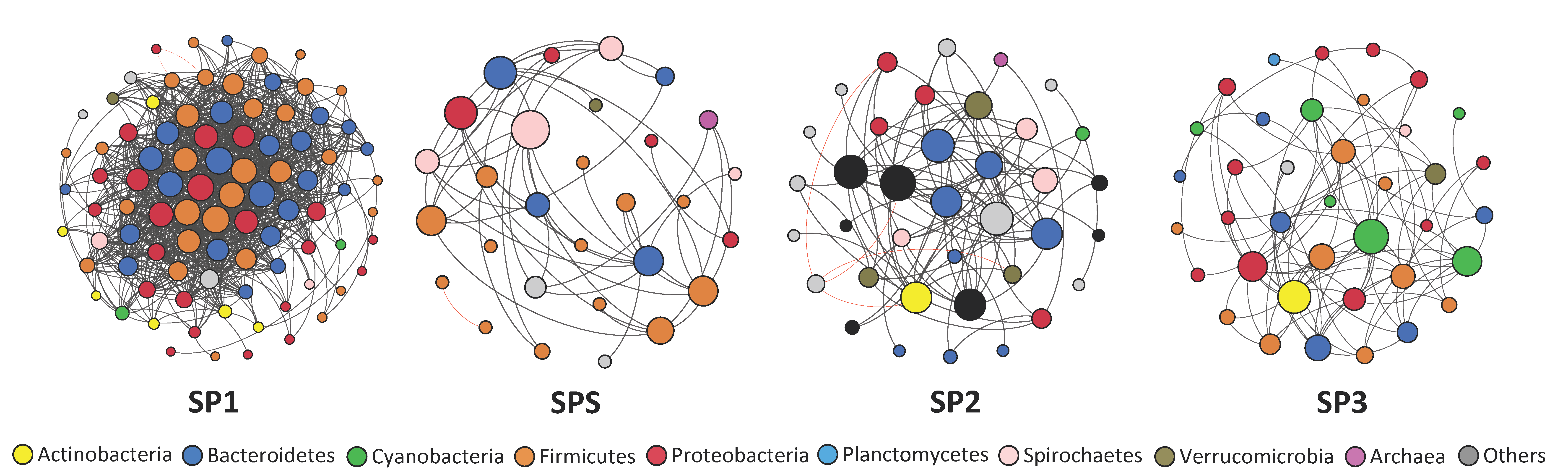
3.5. Co-Occurrence Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WWAP. UN World Water Development Report, Nature-Based Solutions for Water; World Water Assessment Programme: Brasilia, Brazil, 2018. [Google Scholar]
- Rodell, M.; Famiglietti, J.S.; Wiese, D.N.; Reager, J.T.; Beaudoing, H.K.; Landerer, F.W.; Lo, M.-H. Emerging trends in global freshwater availability. Nat. Cell Biol. 2018, 557, 651–659. [Google Scholar] [CrossRef]
- Ait-Kadi, M. Water for Development and Development for Water: Realizing the Sustainable Development Goals (SDGs) Vision. Aquat. Procedia 2016, 6, 106–110. [Google Scholar] [CrossRef]
- Molle, F.; Mollinga, P. Water poverty indicators: Conceptual problems and policy issues. Hydrol. Res. 2003, 5, 529–544. [Google Scholar] [CrossRef]
- Zeeman, G.; Kujawa, K.; de Mes, T.Z.D.; de Graaff, M.S.; Abu-Ghunmi, L.N.A.H.; Mels, A.R.; Meulman, B.; Temmink, B.G.; Buisman, C.J.N.; van Lier, J.; et al. Anaerobic Treatment as a Core Technology for Energy, Nutrients and Water from Source-Separated Domestic Waste(Water). Water Sci. Technol. 2008, 57, 1207–1212. [Google Scholar] [PubMed] [Green Version]
- Kujawa-Roeleveld, K.; Zeeman, G. Anaerobic Treatment in Decentralised and Source-Separation-Based Sanitation Concepts. Rev. Environ. Sci. Bio. Technol. 2006, 5, 115–139. [Google Scholar] [CrossRef]
- Shu, D.; He, Y.; Yue, H.; Wang, Q. Microbial structures and community functions of anaerobic sludge in six full-scale wastewater treatment plants as revealed by 454 high-throughput pyrosequencing. Bioresour. Technol. 2015, 186, 163–172. [Google Scholar] [CrossRef]
- Park, H.-D.; Wells, G.F.; Bae, H.; Criddle, C.S.; Francis, C.A. Occurrence of Ammonia-Oxidizing Archaea in Wastewater Treatment Plant Bioreactors. Appl. Environ. Microbiol. 2006, 72, 5643–5647. [Google Scholar] [CrossRef] [Green Version]
- Nakasaki, K.; Tran, L.T.H.; Idemoto, Y.; Abe, M.; Rollon, A.P. Comparison of organic matter degradation and microbial community during thermophilic composting of two different types of anaerobic sludge. Bioresour. Technol. 2009, 100, 676–682. [Google Scholar] [CrossRef]
- Van Kessel, M.A.H.J.; Stultiens, K.; Slegers, M.F.; Cruz, S.G.; Jetten, M.S.; Kartal, B.; Camp, H.J.O.D. Current perspectives on the application of N-damo and anammox in wastewater treatment. Curr. Opin. Biotechnol. 2018, 50, 222–227. [Google Scholar] [CrossRef]
- Wang, H.; Zeng, Y.; Guo, C.; Bao, Y.; Lu, G.; Reinfelder, J.R.; Dang, Z. Bacterial, archaeal, and fungal community responses to acid mine drainage-laden pollution in a rice paddy soil ecosystem. Sci. Total. Environ. 2018, 107–116. [Google Scholar] [CrossRef]
- Samhan, S.A.; Al-Sa’Ed, R.; Mahmoud, N. Removal of pathogenic microorganisms in pilot?scale UASB?septic tanks and Albireh urban wastewater treatment plant in Palestine. Water Int. 2007, 32, 798–809. [Google Scholar] [CrossRef]
- Medeiros, R.C.; Daniel, L.A.; De Oliveira, G.L.; Hoffmann, M.T. Performance of a small-scale wastewater treatment plant for removal of pathogenic protozoa (oo)cysts and indicator microorganisms. Environ. Technol. 2018, 40, 3492–3501. [Google Scholar] [CrossRef]
- Zhang, L.; De Vrieze, J.; Hendrickx, T.L.; Wei, W.; Temmink, H.; Rijnaarts, H.; Zeeman, G. Anaerobic treatment of raw domestic wastewater in a UASB-digester at 10 °C and microbial community dynamics. Chem. Eng. J. 2018, 334, 2088–2097. [Google Scholar] [CrossRef]
- De Graaff, M.S.; Temmink, H.; Zeeman, G.; Buisman, C.J.N. Anaerobic Treatment of Concentrated Black Water in a UASB Reactor at a Short HRT. Water 2010, 2, 101–119. [Google Scholar] [CrossRef]
- Slompo, N.D.M.; Quartaroli, L.; Fernandes, T.V.; Silva, G.H.R.; Daniel, L.A. Nutrient and pathogen removal from anaerobically treated black water by microalgae. J. Environ. Manag. 2020, 268, 110693. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Friedman, J.; Alm, E. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M. The structure and function of networks. Comput. Phys. Commun. 2002, 147, 40–45. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Net-works. In Proceedings of the International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Chernicharo, C.A.D.L.; Brandt, E.M.F.; Bressani-Ribeiro, T.; Melo, V.R.; Bianchetti, F.J.; MotaFilho, C.R.; McAdam, E. Development of a tool for improving the management of gaseous emissions in UASB-based sewage treatment plants. Water Pract. Technol. 2017, 12, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E. Effect of organic loading rate on the stability, operational parameters and performance of a secondary upflow anaerobic sludge bed reactor treating piggery waste. Bioresour. Technol. 2005, 96, 335–344. [Google Scholar] [CrossRef]
- Lu, X.; Ni, J.; Zhen, G.; Kubota, K.; Cheng, J. Response of morphology and microbial community structure of granules to influent COD/SO42-ratios in an upflow anaerobic sludge blanket (UASB) reactor treating starch wastewater. Bioresour. Technol. 2018, 256, 456–465. [Google Scholar] [CrossRef]
- Ozgun, H.; Gimenez, J.B.; Ersahin, M.E.; Tao, Y.; Spanjers, H.; Van Lier, J.B. Impact of membrane addition for effluent extraction on the performance and sludge characteristics of upflow anaerobic sludge blanket reactors treating municipal wastewater. J. Membr. Sci. 2015, 479, 95–104. [Google Scholar] [CrossRef]
- Syutsubo, K.; Yoochatchaval, W.; Tsushima, I.; Araki, N.; Kubota, K.; Onodera, T.; Takahashi, M.; Yamaguchi, T.; Yoneyama, Y. Evaluation of sludge properties in a pilot-scale UASB reactor for sewage treatment in a temperate region. Water Sci. Technol. 2011, 64, 1959–1966. [Google Scholar] [CrossRef]
- Fernández-Gómez, B.; Richter, M.; Schüler, M.; Pinhassi, J.; Acinas, S.G.; González, J.M.; Pedrós-Alió, C. Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef] [Green Version]
- Rivière, D.; Desvignes, V.; Pelletier, E.; Chaussonnerie, S.; Guermazi, S.; Weissenbach, J.; Li, T.; Camacho, P.; Sghir, A. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J. 2009, 3, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Cardinali-Rezende, J.; Araujo, J.C.; Almeida, P.G.S.; Chernicharo, C.A.L.; Sanz, J.L.; Chartone-Souza, E.; Nascimento, A.M.A. Organic loading rate and food-to-microorganism ratio shape prokaryotic diversity in a demo-scale up-flow anaerobic sludge blanket reactor treating domestic wastewater. Antonie Leeuwenhoek 2013, 104, 993–1003. [Google Scholar] [CrossRef]
- Li, S.; Wu, Z.; Liu, G. Degradation kinetics of toilet paper fiber during wastewater treatment: Effects of solid retention time and microbial community. Chemosphere 2019, 225, 915–926. [Google Scholar] [CrossRef]
- Hattori, S.; Kamagata, Y.; Hanada, S.; Shoun, H. Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 2000, 50, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, L.; Omil, F.; Méndez, R.; Lema, J. Anaerobic hydrolysis and acidogenesis of wastewaters from food industries with high content of organic solids and protein. Water Res. 1999, 33, 3281–3290. [Google Scholar] [CrossRef]
- Stal, L.J. Physiological ecology of cyanobacteria in microbial mats and other communities. New Phytol. 1995, 131, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.P.; Souza, C.L.; Glória, R.M.; Silva, S.Q.; Chernicharo, C.A.L. Biological oxidation of sulphides by microorganisms present in the scum layer of UASB reactors treating domestic wastewater. Water Sci. Technol. 2012, 66, 1871–1878. [Google Scholar] [CrossRef]
- Li, Q.; Wang, X.C.; Zhang, H.; Shi, H.; Hu, T.; Ngo, H. Characteristics of nitrogen transformation and microbial community in an aerobic composting reactor under two typical temperatures. Bioresour. Technol. 2013, 137, 270–277. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Raes, J.; Bogert, B.V.D.; Arumugam, M.; Booijink, C.C.G.M.; Troost, F.J.; Bork, P.; Wels, M.; De Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef]
- Cobo-Díaz, J.F.; Fernández-González, A.J.; Villadas, P.J.; Robles, A.B.; Toro, N.; Fernández-López, M. Metagenomic Assessment of the Potential Microbial Nitrogen Pathways in the Rhizosphere of a Mediterranean Forest After a Wildfire. Microb. Ecol. 2015, 69, 895–904. [Google Scholar] [CrossRef]
- Inoue, J.-I.; Oshima, K.; Suda, W.; Sakamoto, M.; Iino, T.; Noda, S.; Hongoh, Y.; Hattori, M.; Ohkuma, M. Distribution and Evolution of Nitrogen Fixation Genes in the Phylum Bacteroidetes. Microbes Environ. 2015, 30, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Covian, D.; Salazar, N.; Gueimonde, M.; Reyes-Gavilán, C.G.D.L. Shaping the Metabolism of Intestinal Bacteroides Population through Diet to Improve Human Health. Front. Microbiol. 2017, 8, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.M.; Kim, J.H.; Ha, J.H.; Park, J.M. Bacterial and methanogenic archaeal communities during the single-stage anaerobic digestion of high-strength food wastewater. Bioresour. Technol. 2014, 165, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sekiguchi, Y.; Imachi, H.; Kamagata, Y.; Ohashi, A.; Harada, H. Diversity, Localization, and Physiological Properties of Filamentous Microbes Belonging to Chloroflexi Subphylum I in Mesophilic and Thermophilic Methanogenic Sludge Granules. Appl. Environ. Microbiol. 2005, 71, 7493–7503. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Nie, Y.; Kato, H.; Wu, J.; Utashiro, T.; Lu, J.; Yue, S.; Jiang, H.; Zhang, L.; Cheng, J. Methanogenic degradation of toilet-paper cellulose upon sewage treatment in an anaerobic membrane bioreactor at room temperature. Bioresour. Technol. 2017, 228, 69–76. [Google Scholar] [CrossRef]
- Ruiken, C.; Breuer, G.; Klaversma, E.; Santiago, T.; Van Loosdrecht, M. Sieving wastewater-Cellulose recovery, economic and energy evaluation. Water Res. 2013, 47, 43–48. [Google Scholar] [CrossRef]
- Di Termini, I.; Prassone, A.; Cattaneo, C.; Rovatti, M. On the nitrogen and phosphorus removal in algal photobioreactors. Ecol. Eng. 2011, 37, 976–980. [Google Scholar] [CrossRef]
- Gao, F.; Li, C.; Yang, Z.-H.; Zeng, G.-M.; Mu, J.; Liu, M.; Cui, W. Removal of nutrients, organic matter, and metal from domestic secondary effluent through microalgae cultivation in a membrane photobioreactor. J. Chem. Technol. Biotechnol. 2016, 91, 2713–2719. [Google Scholar] [CrossRef]
- Shi, X.; Yeap, T.S.; Huang, S.; Chen, J.; Ng, H.Y. Pretreatment of saline antibiotic wastewater using marine microalga. Bioresour. Technol. 2018, 258, 240–246. [Google Scholar] [CrossRef]
- Suleiman, A.K.A.; Lourenço, K.S.; Clark, C.; Luz, R.L.; Da Silva, G.H.R.; Vet, L.E.; Cantarella, H.; Fernandes, T.V.; Kuramae, E.E. From toilet to agriculture: Fertilization with microalgal biomass from wastewater impacts the soil and rhizosphere active microbiomes, greenhouse gas emissions and plant growth. Resour. Conserv. Recycl. 2020, 161, 104924. [Google Scholar] [CrossRef]
- Aksu, Z. Biosorption of Heavy Metals by Microalgae in Batch and Continuous Systems. In Wastewater Treatment with Algae; Wong, Y.-S., Tam, N.F.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 37–53. [Google Scholar]
- Tchobanoglous, G.; Burton, F.; Stensel, H.D. Wastewater Engineering: Treatment and Reuse; McGraw-Hill Education: New York, NY, USA, 2002. [Google Scholar]
- Delanka-Pedige, H.M.; Munasinghe-Arachchige, S.P.; Cornelius, J.; Henkanatte-Gedera, S.M.; Tchinda, D.; Zhang, Y.; Nirmalakhandan, N. Pathogen reduction in an algal-based wastewater treatment system employing Galdieria sulphuraria. Algal Res. 2019, 39, 101423. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Points of Sampling | CODtotal (g L−1) | TN (g L−1) | TP (g L−1) | Alkalinity (g L−1) | pH | TSS (g L−1) | VSS (g L−1) | T (°C) | DO Mg L−1 |
|---|---|---|---|---|---|---|---|---|---|
| Average ± standard variation | |||||||||
| SP1 | 17.0 ± 4.7 | 2.53 ± 1.89 | 0.20 ± 0.05 | 4.85 ± 1.14 | 8.40 ± 0.30 | 8.2 ± 3.1 | 7.5 ± 3.1 | 18.0 ± 1.8 | 0.58 ± 1.02 |
| SP-S | 43.7 ± 3.4 | 3.85 ± 1.49 | 0.46 ± 0.08 | 5.19 ± 0.10 | 7.60 ± 0.40 | 45.8 ± 0.9 | 36.0 ± 1.2 | 21.6 ± 6.3 | 0.36 ± 0.57 |
| SP2 | 6.7 ± 2.8 | 1.76 ± 1.26 | 0.14 ± 0.02 | 5.19 ± 0.10 | 7.70 ± 0.10 | 4.8 ± 2.4 | 4.6 ± 2.2 | 23.3 ± 9.3 | 0.63 ± 0.71 |
| SP3 | 5.2 ± 2.6 | 1.42 ± 1.21 | 0.07 ± 0.01 | 0.44 ± 0.21 | 6.60 ± 0.40 | 2.8 ± 0.4 | 2.5 ± 0.1 | 22.0 ± 2.6 | 1.10 ± 0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuramae, E.E.; Dimitrov, M.R.; da Silva, G.H.R.; Lucheta, A.R.; Mendes, L.W.; Luz, R.L.; Vet, L.E.M.; Fernandes, T.V. On-Site Blackwater Treatment Fosters Microbial Groups and Functions to Efficiently and Robustly Recover Carbon and Nutrients. Microorganisms 2021, 9, 75. https://doi.org/10.3390/microorganisms9010075
Kuramae EE, Dimitrov MR, da Silva GHR, Lucheta AR, Mendes LW, Luz RL, Vet LEM, Fernandes TV. On-Site Blackwater Treatment Fosters Microbial Groups and Functions to Efficiently and Robustly Recover Carbon and Nutrients. Microorganisms. 2021; 9(1):75. https://doi.org/10.3390/microorganisms9010075
Chicago/Turabian StyleKuramae, Eiko E., Mauricio R. Dimitrov, Gustavo H. R. da Silva, Adriano R. Lucheta, Lucas W. Mendes, Ronildson L. Luz, Louise E. M. Vet, and Tania V. Fernandes. 2021. "On-Site Blackwater Treatment Fosters Microbial Groups and Functions to Efficiently and Robustly Recover Carbon and Nutrients" Microorganisms 9, no. 1: 75. https://doi.org/10.3390/microorganisms9010075