
Aspergillus fumigatus versus Genus Aspergillus: Conservation, Adaptive Evolution and Specific Virulence Genes

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Initial Dataset, Quality Assessment and Filtering
2.2. Orthologs and Unique Genes
2.3. Reconciling of Gene Trees and Species Trees
2.4. Single-Copy Orthologs Gene Families Data Set
2.5. Multiple Sequence Alignment (MSA)
2.6. Test for Recombination
2.7. Positive Selection in A. fumigatus Protein-Coding Genes
2.8. Tests of Functional Category Enrichment
2.9. Interolog Network
2.10. Inference of Positive Selection in A. fumigatus Virulence Genes
2.11. Statistical Analysis of PSGs
3. Results
3.1. Genomic Features and Quality Assessment
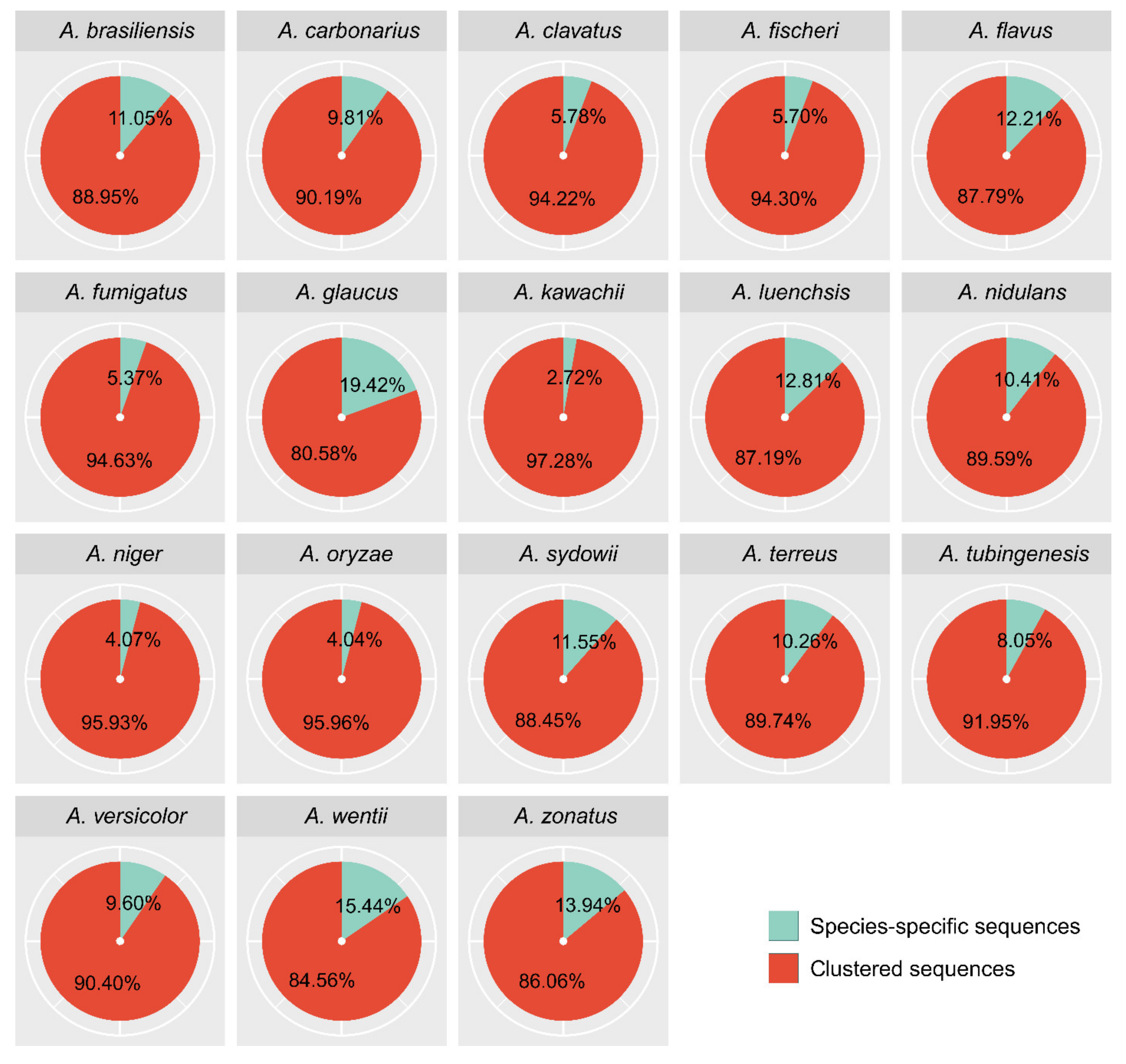
3.2. Orthologs and Unique Genes
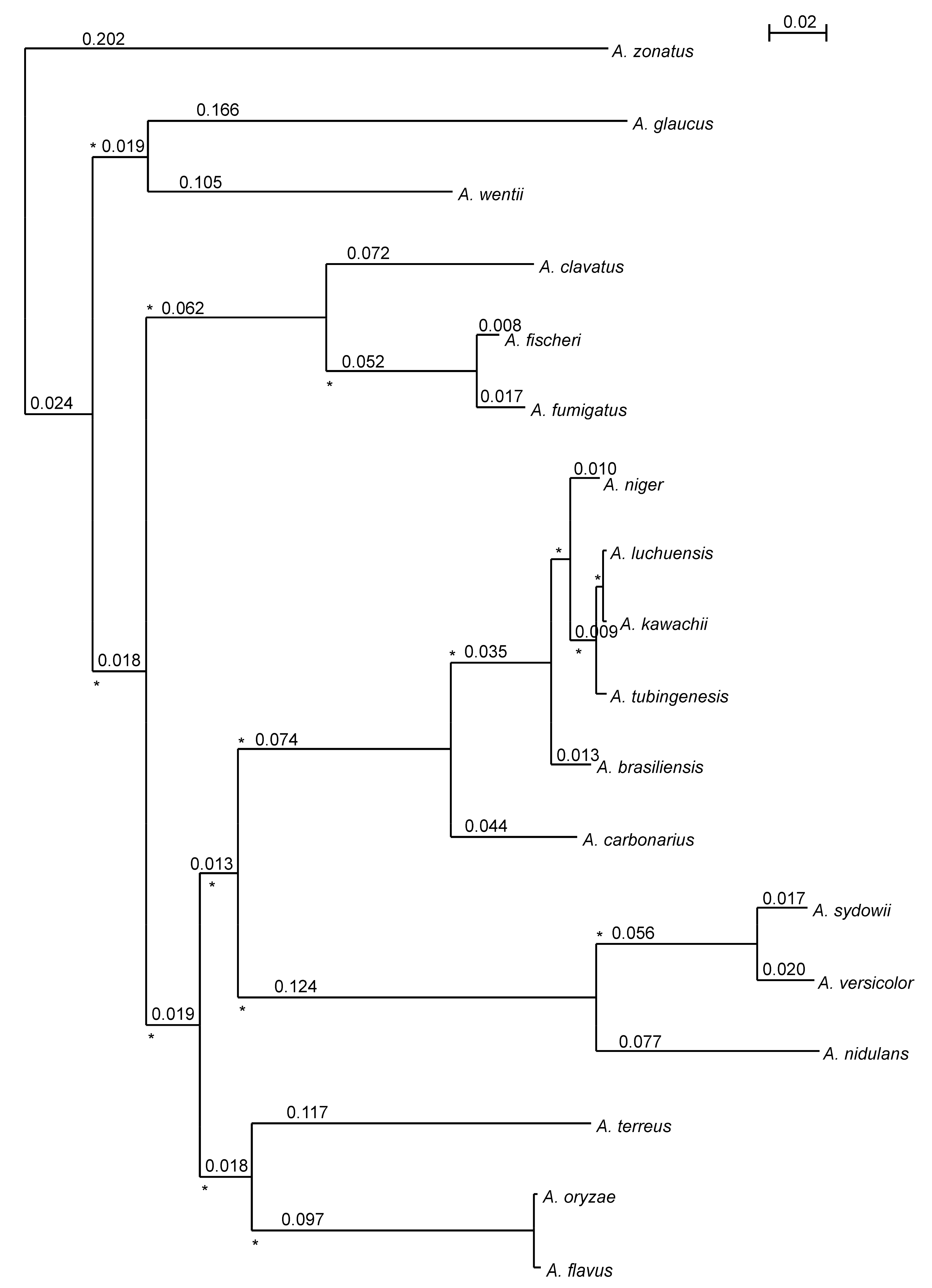
3.3. Reconciling of Species Tree and Gene Trees
3.4. Positive Selection in A. fumigatus Single-Copy Ortholog Protein-Coding Genes
3.5. Host-Interacting Single-Copy Ortholog PSGs
3.6. Functional Classification of Single-Copy Ortholog PSGs
3.7. Positive Selection in A. fumigatus in Multi-Gene Families
3.8. Ancient Defenses: Example Gliotoxin Biosynthesis Cluster
4. Discussion
4.1. Species-Specific A. fumigatus Proteins
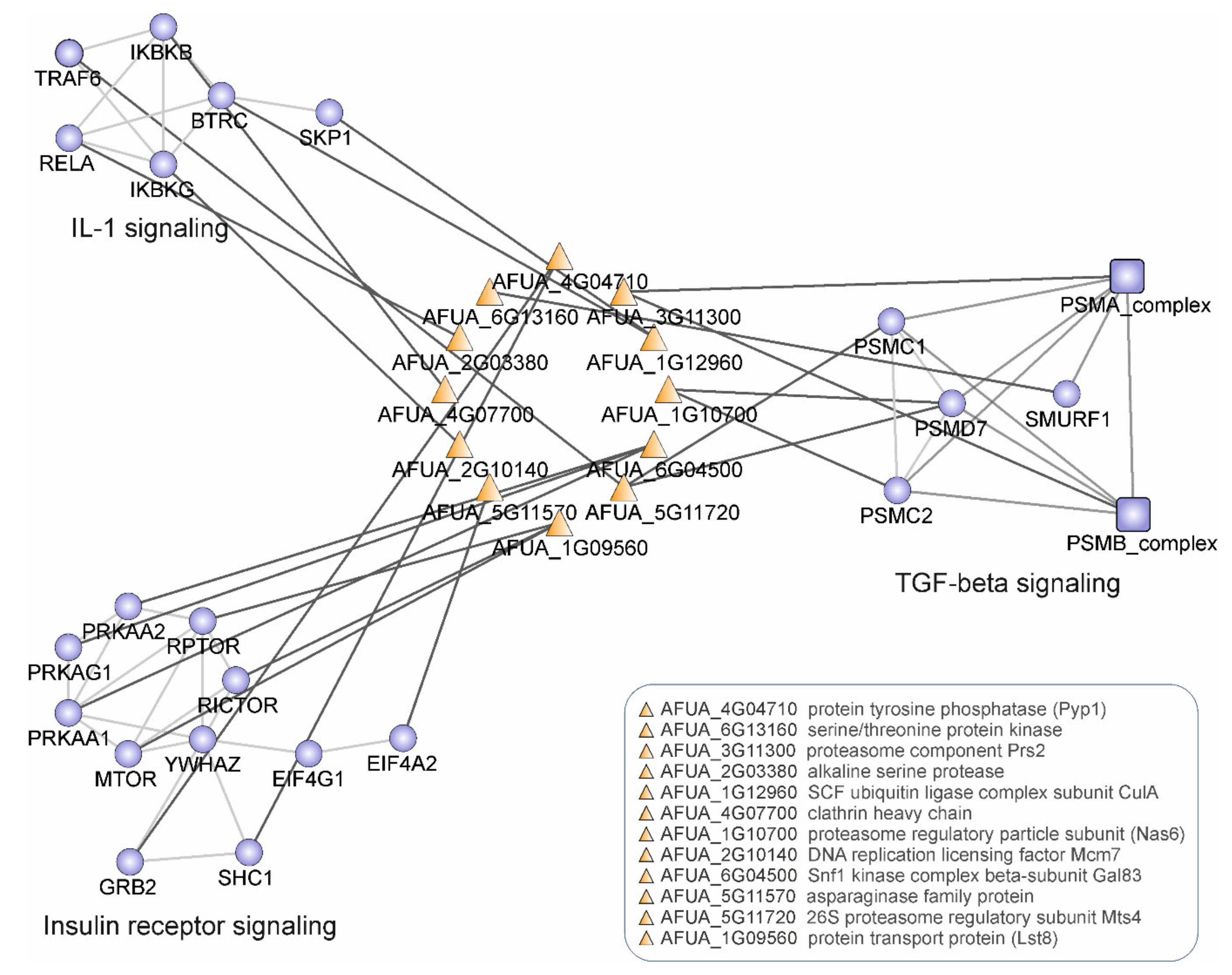
4.2. Pathway and GO Over-Representation of A. fumigatus SCO Proteins Interacting with Human Host Proteins
4.3. Positive Selection in Other Functionally Annotated Categories
4.3.1. Conserved Hypothetical Protein-Coding Genes
4.3.2. Hypoxia-Responsive Genes
4.3.3. Genes Involved in Early Development of A. fumigatus
4.3.4. Essential Protein-Coding Genes
4.4. Positive Selection in A. fischeri Protein-Coding Genes
4.5. A. fumigatus Multi-Copy Virulence Genes with Positive Selection
4.6. Evolutionary Overview
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Basturkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef]
- Sugui, J.A.; Kwon-Chung, K.J.; Juvvadi, P.R.; Latge, J.P.; Steinbach, W.J. Aspergillus fumigatus and related species. Cold Spring Harb. Perspect. Med. 2014, 5, a019786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, C.S.; Ho, P.L.; Yuen, K.Y. Simultaneous Aspergillus fischeri and Herpes simplex pneumonia in a patient with multiple myeloma. Scand. J. Infect. Dis. 1998, 30, 190–191. [Google Scholar]
- Gerber, J.; Chomicki, J.; Brandsberg, J.W.; Jones, R.; Hammerman, K.J. Pulmonary aspergillosis caused by Aspergillus fischeri var. spinosus: Report of a case and value of serologic studies. Am. J. Clin. Pathol. 1973, 60, 861–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, M.E.; Knowles, S.L.; Raja, H.A.; Beattie, S.R.; Kowalski, C.H.; Steenwyk, J.L.; Silva, L.P.; Chiaratto, J.; Ries, L.N.A.; Goldman, G.H.; et al. Characterizing the pathogenic, genomic, and chemical traits of Aspergillus fischeri, a close relative of the major human fungal pathogen Aspergillus fumigatus. Msphere 2019, 4, e00018-19. [Google Scholar] [CrossRef] [Green Version]
- Houbraken, J.; Weig, M.; Gross, U.; Meijer, M.; Bader, O. Aspergillus oerlinghausenensis, a new mould species closely related to A. fumigatus. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [Green Version]
- De Vries, R.P.; Riley, R.; Wiebenga, A.; Aguilar-Osorio, G.; Amillis, S.; Uchima, C.A.; Anderluh, G.; Asadollahi, M.; Askin, M.; Barry, K.; et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 2017, 18, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Gladieux, P.; Ropars, J.; Badouin, H.; Branca, A.; Aguileta, G.; de Vienne, D.M.; Rodriguez de la Vega, R.C.; Branco, S.; Giraud, T. Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 2014, 23, 753–773. [Google Scholar] [CrossRef]
- Winkler, J.; Kao, K.C. Harnessing recombination to speed adaptive evolution in Escherichia coli. Metab. Eng. 2012, 14, 487–495. [Google Scholar] [CrossRef]
- Shapiro, B.J.; David, L.A.; Friedman, J.; Alm, E.J. Looking for Darwin’s footprints in the microbial world. Trends Microbiol. 2009, 17, 196–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacColl, A.D. The ecological causes of evolution. Trends Ecol. Evol. 2011, 26, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.G.; Rokas, A. The function and evolution of the Aspergillus genome. Trends Microbiol. 2013, 21, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Casadevall, A.; Pirofski, L.A. Accidental virulence, cryptic pathogenesis, martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryot Cell 2007, 6, 2169–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillmann, F.; Novohradska, S.; Mattern, D.J.; Forberger, T.; Heinekamp, T.; Westermann, M.; Winckler, T.; Brakhage, A.A. Virulence determinants of the human pathogenic fungus Aspergillus fumigatus protect against soil amoeba predation. Environ. Microbiol. 2015, 17, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, N.D.; Khaldi, N.; Joardar, V.S.; Maiti, R.; Amedeo, P.; Anderson, M.J.; Crabtree, J.; Silva, J.C.; Badger, J.H.; Albarraq, A.; et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008, 4, e1000046. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2005, 33, D34–D38. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Hambuch, T.M.; Parsch, J. Patterns of synonymous codon usage in Drosophila melanogaster genes with sex-biased expression. Genetics 2005, 170, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- De Queiroz, A.; Gatesy, J. The supermatrix approach to systematics. Trends Ecol. Evol. 2007, 22, 34–41. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Diekmann, Y.; Pereira-Leal, J.B. Gene tree affects inference of sites under selection by the branch-site test of positive selection. Evol. Bioinform. Online 2015, 11, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Philippe, H.; Lopez, P.; Brinkmann, H.; Budin, K.; Germot, A.; Laurent, J.; Moreira, D.; Muller, M.; Le Guyader, H. Early-branching or fast-evolving eukaryotes? An answer based on slowly evolving positions. Proc. Biol. Sci. 2000, 267, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Hagelsieb, G.; Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 2008, 24, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, S.; Abreu-Goodger, C. Using MCL to extract clusters from networks. Methods Mol. Biol. 2012, 804, 281–295. [Google Scholar]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Jordan, G.; Goldman, N. The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol. Biol. Evol. 2012, 29, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Privman, E.; Penn, O.; Pupko, T. Improving the performance of positive selection inference by filtering unreliable alignment regions. Mol. Biol. Evol. 2012, 29, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, W.; Yang, Z. The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol. Biol. Evol. 2010, 27, 2257–2267. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; dos Reis, M. Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 2011, 28, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Self, S.G.; Liang, K.-Y. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J. Am. Stat. Assoc. 1987, 82, 605–610. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host-pathogen interactions. Database 2016, 2016. [Google Scholar] [CrossRef]
- Durmus Tekir, S.; Cakir, T.; Ardic, E.; Sayilirbas, A.S.; Konuk, G.; Konuk, M.; Sariyer, H.; Ugurlu, A.; Karadeniz, I.; Ozgur, A.; et al. PHISTO: Pathogen-host interaction search tool. Bioinformatics 2013, 29, 1357–1358. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Luscombe, N.M.; Lu, H.X.; Zhu, X.; Xia, Y.; Han, J.D.; Bertin, N.; Chung, S.; Vidal, M.; Gerstein, M. Annotation transfer between genomes: Protein-protein interologs and protein-DNA regulogs. Genome Res. 2004, 14, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Sonnhammer, E.L.; Ostlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, D234–D239. [Google Scholar] [CrossRef]
- Gupta, S.K.; Osmanoglu, Ö.; Srivastava, M.; Bencúrová, E.; Dandekar, T. Pathogen and host-pathogen protein interactions provide a key to identify novel drug targets. In Reference Module in Biomedical Sciences; Systems Medicine: Integrative, Qualitative and Computational Approaches; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Turner, H. A peer-reviewed, open-access publication of the R Foundation for Statistical Computing. R J. 2011, 3, 3. [Google Scholar]
- Okagaki, L.H.; Sailsbery, J.K.; Eyre, A.W.; Dean, R.A. Comparative genome analysis and genome evolution of members of the magnaporthaceae family of fungi. BMC Genom. 2016, 17, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, H. Predicting secretory proteins with SignalP. Methods Mol. Biol. 2017, 1611, 59–73. [Google Scholar] [PubMed] [Green Version]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Gee, H. Evolution: Ending incongruence. Nature 2003, 425, 782. [Google Scholar] [CrossRef]
- Riddle, H.F.; Channell, S.; Blyth, W.; Weir, D.M.; Lloyd, M.; Amos, W.M.; Grant, I.W. Allergic alveolitis in a maltworker. Thorax 1968, 23, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Nicod, C.; Banaei-Esfahani, A.; Collins, B.C. Elucidation of host-pathogen protein-protein interactions to uncover mechanisms of host cell rewiring. Curr. Opin. Microbiol. 2017, 39, 7–15. [Google Scholar] [CrossRef]
- Remmele, C.W.; Luther, C.H.; Balkenhol, J.; Dandekar, T.; Muller, T.; Dittrich, M.T. Integrated inference and evaluation of host-fungi interaction networks. Front. Microbiol. 2015, 6, 764. [Google Scholar] [CrossRef] [Green Version]
- Hohl, T.M.; Feldmesser, M. Aspergillusfumigatus: Principles of pathogenesis and host defense. Eukaryot. Cell 2007, 6, 1953–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertolas-Balint, F.; Rossen, J.W.A.; Oliveira Dos Santos, C.; Chlebowicz, M.M.A.; Raangs, E.C.; van Putten, M.L.; Sola-Campoy, P.J.; Han, L.; Schmidt, M.; Garcia-Cobos, S. Revealing the virulence potential of clinical and environmental Aspergillus fumigatus isolates using whole-genome sequencing. Front. Microbiol. 2019, 10, 1970. [Google Scholar] [CrossRef] [Green Version]
- Vivek-Ananth, R.P.; Mohanraj, K.; Vandanashree, M.; Jhingran, A.; Craig, J.P.; Samal, A. Comparative systems analysis of the secretome of the opportunistic pathogen Aspergillus fumigatus and other Aspergillus species. Sci. Rep. 2018, 8, 6617. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases 2014, 5, e983863. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Bidochka, M.J.; Clark, D.C.; Lewis, M.W.; Keyhani, N.O. Could insect phagocytic avoidance by entomogenous fungi have evolved via selection against soil amoeboid predators? Microbiology 2010, 156, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Novohradska, S.; Ferling, I.; Hillmann, F. Exploring virulence determinants of filamentous fungal pathogens through interactions with soil amoebae. Front. Cell Infect. Microbiol 2017, 7, 497. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.L.; Mead, M.E.; Silva, L.P.; Raja, H.A.; Steenwyk, J.L.; Goldman, G.H.; Oberlies, N.H.; Rokas, A. Gliotoxin, a known virulence factor in the major human pathogen Aspergillus fumigatus, is also biosynthesized by its nonpathogenic relative Aspergillus fischeri. mBio 2020, 11, e03361-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duret, L. Neutral theory: The null hypothesis of molecular evolution. Nat. Educ. 2008, 1, 803–806. [Google Scholar]
- Van de Veerdonk, F.L.; Gresnigt, M.S.; Romani, L.; Netea, M.G.; Latge, J.P. Aspergillusfumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 2017, 15, 661–674. [Google Scholar] [CrossRef]
- Carbone, I.; Jakobek, J.L.; Ramirez-Prado, J.H.; Horn, B.W. Recombination, balancing selection and adaptive evolution in the aflatoxin gene cluster of Aspergillus parasiticus. Mol. Ecol. 2007, 16, 4401–4417. [Google Scholar] [CrossRef]
- Yang, E.; Hulse, A.M.; Cai, J.J. Evolutionary analysis of sequence divergence and diversity of duplicate genes in Aspergillus fumigatus. Evol. Bioinform. Online 2012, 8, 623–644. [Google Scholar] [CrossRef]
- Wu, C.I.; Ting, C.T. Genes and speciation. Nat. Rev. Genet. 2004, 5, 114–122. [Google Scholar] [CrossRef]
- Nielsen, R.; Bustamante, C.; Clark, A.G.; Glanowski, S.; Sackton, T.B.; Hubisz, M.J.; Fledel-Alon, A.; Tanenbaum, D.M.; Civello, D.; White, T.J.; et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005, 3, e170. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zhang, G.; Zhang, Y.; Xu, S.; Zhao, R.; Zhan, Z.; Li, X.; Ding, Y.; Yang, S.; Wang, W. On the origin of new genes in Drosophila. Genome Res. 2008, 18, 1446–1455. [Google Scholar] [CrossRef] [Green Version]
- Letterio, J.J.; Roberts, A.B. Regulation of immune responses by TGF-beta. Annu Rev. Immunol 1998, 16, 137–161. [Google Scholar] [CrossRef] [Green Version]
- Karpac, J.; Younger, A.; Jasper, H. Dynamic coordination of innate immune signaling and insulin signaling regulates systemic responses to localized DNA damage. Dev. Cell 2011, 20, 841–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balloy, V.; Chignard, M. The innate immune response to Aspergillus fumigatus. Microbes Infect. 2009, 11, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Croston, T.L.; Nayak, A.P.; Lemons, A.R.; Goldsmith, W.T.; Gu, J.K.; Germolec, D.R.; Beezhold, D.H.; Green, B.J. Influence of Aspergillus fumigatus conidia viability on murine pulmonary microRNA and mRNA expression following subchronic inhalation exposure. Clin. Exp. Allergy 2016, 46, 1315–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarsaikhan, N.; Tsoggerel, A.; Hug, C.; Templeton, S.P. The Metabolic cytokine adiponectin inhibits inflammatory lung pathology in invasive aspergillosis. J. Immunol. 2019, 203, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.D.; Ayubi, T.; Chung, D.; Tubau-Juni, N.; Leber, A.; Dang, H.X.; Karyala, S.; Hontecillas, R.; Lawrence, C.B.; Cramer, R.A.; et al. Modulation of immune signaling and metabolism highlights host and fungal transcriptional responses in mouse models of invasive pulmonary aspergillosis. Sci. Rep. 2017, 7, 17096. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, A.K.; Lehmann, M.M.; Zickovich, J.M.; Espinosa, V.; Shepardson, K.M.; Watschke, C.P.; Hilmer, K.M.; Thammahong, A.; Barker, B.M.; Rivera, A.; et al. IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog. 2015, 11, e1004625. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, A.; Insall, R.H. Small GTPases in Dictyostelium: Lessons from a social amoeba. Trends Genet. 2001, 17, 41–48. [Google Scholar] [CrossRef]
- Swanson, J.A. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008, 9, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Kruppa, A.J.; Kendrick-Jones, J.; Buss, F. Myosins, actin and autophagy. Traffic 2016, 17, 878–890. [Google Scholar] [CrossRef] [Green Version]
- Duhon, D.; Cardelli, J. The regulation of phagosome maturation in Dictyostelium. J. Muscle Res. Cell Motil. 2002, 23, 803–808. [Google Scholar] [CrossRef]
- Steenbergen, J.N.; Shuman, H.A.; Casadevall, A. Cryptococcus neoformans interactions with amoebae suggest an explanation for its virulence and intracellular pathogenic strategy in macrophages. Proc. Natl. Acad. Sci. USA 2001, 98, 15245–15250. [Google Scholar] [CrossRef] [Green Version]
- Galperin, M.Y.; Koonin, E.V. ‘Conserved hypothetical’ proteins: Prioritization of targets for experimental study. Nucleic Acids Res. 2004, 32, 5452–5463. [Google Scholar] [CrossRef] [Green Version]
- Kroll, K.; Pahtz, V.; Hillmann, F.; Vaknin, Y.; Schmidt-Heck, W.; Roth, M.; Jacobsen, I.D.; Osherov, N.; Brakhage, A.A.; Kniemeyer, O. Identification of hypoxia-inducible target genes of Aspergillus fumigatus by transcriptome analysis reveals cellular respiration as an important contributor to hypoxic survival. Eukaryot Cell 2014, 13, 1241–1253. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Sznajder, J.I. Effects of hypoxia on the alveolar epithelium. Proc. Am. Thorac. Soc. 2005, 2, 202–205. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Lee, J.A.; Underwood, J.C.; Harris, A.L.; Lewis, C.E. Macrophage responses to hypoxia: Relevance to disease mechanisms. J. Leukoc. Biol. 1999, 66, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Lamarre, C.; Sokol, S.; Debeaupuis, J.P.; Henry, C.; Lacroix, C.; Glaser, P.; Coppee, J.Y.; Francois, J.M.; Latge, J.P. Transcriptomic analysis of the exit from dormancy of Aspergillus fumigatus conidia. BMC Genom. 2008, 9, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagas, S.E.; Jain, M.R.; Li, H.; Perlin, D.S. The proteomic signature of Aspergillus fumigatus during early development. Mol. Cell Proteom. 2011, 10, M111010108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, D.; Suzuki, S.; Kamei, K.; Gonoi, T.; Kawamoto, S. The role of AtfA and HOG MAPK pathway in stress tolerance in conidia of Aspergillus fumigatus. Fungal Genet. Biol. 2014, 73, 138–149. [Google Scholar] [CrossRef]
- Hagiwara, D.; Takahashi, H.; Kusuya, Y.; Kawamoto, S.; Kamei, K.; Gonoi, T. Comparative transcriptome analysis revealing dormant conidia and germination associated genes in Aspergillus species: An essential role for AtfA in conidial dormancy. BMC Genom. 2016, 17, 358. [Google Scholar] [CrossRef] [Green Version]
- Van Zeebroeck, G.; Kimpe, M.; Vandormael, P.; Thevelein, J.M. A split-ubiquitin two-hybrid screen for proteins physically interacting with the yeast amino acid transceptor Gap1 and ammonium transceptor Mep2. PLoS ONE 2011, 6, e24275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joardar, V.; Abrams, N.F.; Hostetler, J.; Paukstelis, P.J.; Pakala, S.; Pakala, S.B.; Zafar, N.; Abolude, O.O.; Payne, G.; Andrianopoulos, A.; et al. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genom. 2012, 13, 698. [Google Scholar] [CrossRef]
- Hunt, M.C.; Siponen, M.I.; Alexson, S.E. The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. Biochim. Biophys. Acta 2012, 1822, 1397–1410. [Google Scholar] [CrossRef] [Green Version]
- Moffat, C.; Bhatia, L.; Nguyen, T.; Lynch, P.; Wang, M.; Wang, D.; Ilkayeva, O.R.; Han, X.; Hirschey, M.D.; Claypool, S.M.; et al. Acyl-CoA thioesterase-2 facilitates mitochondrial fatty acid oxidation in the liver. J. Lipid Res. 2014, 55, 2458–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, O.; Sari, F.; Braus, G.H.; Irniger, S. The protein kinase ImeB is required for light-mediated inhibition of sexual development and for mycotoxin production in Aspergillus nidulans. Mol. Microbiol. 2009, 71, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 2003, 1, 127–136. [Google Scholar] [CrossRef]
- Jordan, I.K.; Rogozin, I.B.; Wolf, Y.I.; Koonin, E.V. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002, 12, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Gao, F.; Lin, Y. Evolutionary conservation analysis between the essential and nonessential genes in bacterial genomes. Sci Rep. 2015, 5, 13210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, G.; Rocha, E.; Danchin, A. How essential are nonessential genes? Mol. Biol. Evol. 2005, 22, 2147–2156. [Google Scholar] [CrossRef]
- Thykaer, J.; Andersen, M.R.; Baker, S.E. Essential pathway identification: From in silico analysis to potential antifungal targets in Aspergillus fumigatus. Med. Mycol. 2009, 47, S80–S87. [Google Scholar] [CrossRef]
- Nierman, W.C.; Pain, A.; Anderson, M.J.; Wortman, J.R.; Kim, H.S.; Arroyo, J.; Berriman, M.; Abe, K.; Archer, D.B.; Bermejo, C.; et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 2005, 438, 1151–1156. [Google Scholar] [CrossRef]
- Lu, Y.; Deng, J.; Rhodes, J.C.; Lu, H.; Lu, L.J. Predicting essential genes for identifying potential drug targets in Aspergillus fumigatus. Comput. Biol. Chem. 2014, 50, 29–40. [Google Scholar] [CrossRef]
- Hu, W.; Sillaots, S.; Lemieux, S.; Davison, J.; Kauffman, S.; Breton, A.; Linteau, A.; Xin, C.; Bowman, J.; Becker, J.; et al. Essential gene identification and drug target prioritization in Aspergillus fumigatus. PLoS Pathog. 2007, 3, e24. [Google Scholar] [CrossRef]
- Carr, P.D.; Tuckwell, D.; Hey, P.M.; Simon, L.; d’Enfert, C.; Birch, M.; Oliver, J.D.; Bromley, M.J. The transposon impala is activated by low temperatures: Use of a controlled transposition system to identify genes critical for viability of Aspergillus fumigatus. Eukaryot Cell 2010, 9, 438–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.M.; Korbel, J.O.; Gerstein, M.B. Positive selection at the protein network periphery: Evaluation in terms of structural constraints and cellular context. Proc. Natl. Acad. Sci. USA 2007, 104, 20274–20279. [Google Scholar] [CrossRef] [Green Version]
- Fraser, H.B.; Hirsh, A.E.; Steinmetz, L.M.; Scharfe, C.; Feldman, M.W. Evolutionary rate in the protein interaction network. Science 2002, 296, 750–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaltdorf, M.; Srivastava, M.; Gupta, S.K.; Liang, C.; Binder, J.; Dietl, A.M.; Meir, Z.; Haas, H.; Osherov, N.; Krappmann, S.; et al. Systematic identification of anti-fungal drug targets by a metabolic network approach. Front. Mol. Biosci. 2016, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crua Asensio, N.; Munoz Giner, E.; de Groot, N.S.; Torrent Burgas, M. Centrality in the host-pathogen interactome is associated with pathogen fitness during infection. Nat. Commun. 2017, 8, 14092. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Goddard, M.R.; Godfray, H.C.; Burt, A. Sex increases the efficacy of natural selection in experimental yeast populations. Nature 2005, 434, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Dasari, P.; Shopova, I.A.; Stroe, M.; Wartenberg, D.; Martin-Dahse, H.; Beyersdorf, N.; Hortschansky, P.; Dietrich, S.; Cseresnyes, Z.; Figge, M.T.; et al. Aspf2 from Aspergillus fumigatus recruits human immune regulators for immune evasion and cell damage. Front. Immunol. 2018, 9, 1635. [Google Scholar] [CrossRef] [Green Version]
- Schrettl, M.; Ibrahim-Granet, O.; Droin, S.; Huerre, M.; Latge, J.P.; Haas, H. The crucial role of the Aspergillus fumigatus siderophore system in interaction with alveolar macrophages. Microbes Infect. 2010, 12, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Van Waeyenberghe, L.; Bare, J.; Pasmans, F.; Claeys, M.; Bert, W.; Haesebrouck, F.; Houf, K.; Martel, A. Interaction of Aspergillus fumigatus conidia with Acanthamoeba castellanii parallels macrophage-fungus interactions. Environ. Microbiol. Rep. 2013, 5, 819–824. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.; Fedorova, N.D.; Crabtree, J.; Yu, Y.; Kim, S.; Chen, D.; Loss, O.; Cairns, T.; Goldman, G.; Armstrong-James, D.; et al. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog. 2008, 4, e1000154. [Google Scholar] [CrossRef]
- Slater, J.L.; Gregson, L.; Denning, D.W.; Warn, P.A. Pathogenicity of Aspergillus fumigatus mutants assessed in Galleria mellonella matches that in mice. Med. Mycol. 2011, 49, S107–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinchen, W.; Lackner, G.; Yasmin, S.; Schrettl, M.; Dahse, H.M.; Haas, H.; Hoffmeister, D. Bimodular peptide synthetase SidE produces fumarylalanine in the human pathogen Aspergillus fumigatus. Appl. Environ. Microbiol. 2013, 79, 6670–6676. [Google Scholar] [CrossRef] [Green Version]
- Bruns, S.; Seidler, M.; Albrecht, D.; Salvenmoser, S.; Remme, N.; Hertweck, C.; Brakhage, A.A.; Kniemeyer, O.; Muller, F.M. Functional genomic profiling of Aspergillus fumigatus biofilm reveals enhanced production of the mycotoxin gliotoxin. Proteomics 2010, 10, 3097–3107. [Google Scholar] [CrossRef]
- Carberry, S.; Molloy, E.; Hammel, S.; O’Keeffe, G.; Jones, G.W.; Kavanagh, K.; Doyle, S. Gliotoxin effects on fungal growth: Mechanisms and exploitation. Fungal Genet. Biol. 2012, 49, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Lambou, K.; Lamarre, C.; Beau, R.; Dufour, N.; Latge, J.P. Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol. Microbiol. 2010, 75, 910–923. [Google Scholar] [CrossRef]
- Oberegger, H.; Zadra, I.; Schoeser, M.; Haas, H. Iron starvation leads to increased expression of Cu/Zn-superoxide dismutase in Aspergillus. FEBS Lett. 2000, 485, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Holdom, M.D.; Lechenne, B.; Hay, R.J.; Hamilton, A.J.; Monod, M. Production and characterization of recombinant Aspergillus fumigatus Cu, Zn superoxide dismutase and its recognition by immune human sera. J. Clin. Microbiol. 2000, 38, 558–562. [Google Scholar] [CrossRef] [Green Version]
- Meneau, I.; Coste, A.T.; Sanglard, D. Identification of Aspergillus fumigatus multidrug transporter genes and their potential involvement in antifungal resistance. Med. Mycol. 2016, 54, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Abad, A.; Fernandez-Molina, J.V.; Bikandi, J.; Ramirez, A.; Margareto, J.; Sendino, J.; Hernando, F.L.; Ponton, J.; Garaizar, J.; Rementeria, A. What makes Aspergillus fumigatus a successful pathogen? Genes and molecules involved in invasive aspergillosis. Rev. Iberoam. Micol. 2010, 27, 155–182. [Google Scholar] [CrossRef]
- Paul, S.; Diekema, D.; Moye-Rowley, W.S. Contributions of Aspergillus fumigatus ATP-binding cassette transporter proteins to drug resistance and virulence. Eukaryot Cell 2013, 12, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroe, M.C.; Netzker, T.; Scherlach, K.; Kruger, T.; Hertweck, C.; Valiante, V.; Brakhage, A.A. Targeted induction of a silent fungal gene cluster encoding the bacteria-specific germination inhibitor fumigermin. Elife 2020, 9, e52541. [Google Scholar] [CrossRef]
- Marcos, C.M.; de Oliveira, H.C.; de Melo, W.C.; da Silva, J.F.; Assato, P.A.; Scorzoni, L.; Rossi, S.A.; de Paula, E.S.A.C.; Mendes-Giannini, M.J.; Fusco-Almeida, A.M. Anti-immune strategies of pathogenic fungi. Front. Cell Infect. Microbiol. 2016, 6, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulussen, C.; Hallsworth, J.E.; Alvarez-Perez, S.; Nierman, W.C.; Hamill, P.G.; Blain, D.; Rediers, H.; Lievens, B. Ecology of aspergillosis: Insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microb. Biotechnol. 2017, 10, 296–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loussert, C.; Schmitt, C.; Prevost, M.C.; Balloy, V.; Fadel, E.; Philippe, B.; Kauffmann-Lacroix, C.; Latge, J.P.; Beauvais, A. In vivo biofilm composition of Aspergillus fumigatus. Cell Microbiol. 2010, 12, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Rank, C.; Klejnstrup, M.L.; Petersen, L.M.; Kildgaard, S.; Frisvad, J.C.; Held Gotfredsen, C.; Ostenfeld Larsen, T. Comparative Chemistry of Aspergillus oryzae (RIB40) and A. flavus (NRRL 3357). Metabolites 2012, 2, 39–56. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Name | Annotation | Category |
|---|---|---|---|
| Afua_4g09580 | aspf2 | Allergen Asp f2 | Allergens |
| Afua_5g03520 | Immunoreactive secreted protein | ||
| Afua_3g03420 | sidD | Nonribosomal peptide synthetase 4 | Nutrient uptake |
| Afua_1g10390 | abcB | Putative ABC multidrug transporter | Resistance to immune response |
| Afua_1g17440 | abcA | ABC drug exporter | |
| Afua_5g09240 | sodA | Cu/Zn superoxide dismutase | |
| Afua_7g00480 | abcE | Putative ABC transporter | |
| Afua_2g00660 | tcsB | Putative sensor histidine kinase/response regulator | Signaling and regulation |
| Afua_2g18060 | fgaMT | 4-dimethylallyltryptophan N-methyltransferase | Toxins and secondary metabolites |
| Afua_4g14490 | tpcJ | Putative dihydrogeodin oxidase | |
| Afua_8g00370 | fma-PKS | Fumagillin biosynthesis polyketide synthase | |
| Afua_8g00440 | Dual-functional monooxygenase/methyltransferase | ||
| Afua_8g00460 | Methionine aminopeptidase type I, putative |
| Gene | Gene Name | Annotation | q-Value (BY) |
|---|---|---|---|
| Afua_6g09660 | gliP | cyclo (L-Phe-L-Ser) synthetase | 1 |
| Afua_6g09670 | gliC | cyclo(L-Phe-L-Ser) hydroxylase | 1 |
| Afua_6g09690 | gliG | glutathione S-transferase | 1 |
| Afua_6g09700 | gliK | gamma-glutamylcyclotransferase | 0.16 |
| Afua_6g09650 | gliJ | 3-benzyl-3,6-bis(cysteinylglycine)-6-(hydroxymethyl)-diketopiperazine dipeptidase | 0.42 |
| Afua_6g09640 | glil | 3-benzyl-3,6-bis(cysteinyl)-6-(hydroxymethyl)-diketopiperazine lyase | 0.16 |
| Afua_6g09740 | gliT | 3-benzyl-3,6-dithio-6-(hydroxymethyl)-diketopiperazine oxidase | 1 |
| Afua_6g09720 | gliN | N-desmethyl-gliotoxin N-methyltransferase | -- * |
| Afua_6g09630 | gliZ | Zn2Cys6 binuclear transcription factor | -- * |
| Afua_6g09680 | gliM | Predicted O-methyltransferase | 1 |
| Afua_6g09710 | gliA | Predicted major facilitator type glioxin transporter | 1 |
| Afua_6g09730 | gliF | Predicted cytochrome P450 monooxygenase | 0.53 |
| Afua_6g09750 | gliH | conserved hypothetical protein | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.K.; Srivastava, M.; Osmanoglu, Ö.; Xu, Z.; Brakhage, A.A.; Dandekar, T. Aspergillus fumigatus versus Genus Aspergillus: Conservation, Adaptive Evolution and Specific Virulence Genes. Microorganisms 2021, 9, 2014. https://doi.org/10.3390/microorganisms9102014
Gupta SK, Srivastava M, Osmanoglu Ö, Xu Z, Brakhage AA, Dandekar T. Aspergillus fumigatus versus Genus Aspergillus: Conservation, Adaptive Evolution and Specific Virulence Genes. Microorganisms. 2021; 9(10):2014. https://doi.org/10.3390/microorganisms9102014
Chicago/Turabian StyleGupta, Shishir K., Mugdha Srivastava, Özge Osmanoglu, Zhuofei Xu, Axel A. Brakhage, and Thomas Dandekar. 2021. "Aspergillus fumigatus versus Genus Aspergillus: Conservation, Adaptive Evolution and Specific Virulence Genes" Microorganisms 9, no. 10: 2014. https://doi.org/10.3390/microorganisms9102014
APA StyleGupta, S. K., Srivastava, M., Osmanoglu, Ö., Xu, Z., Brakhage, A. A., & Dandekar, T. (2021). Aspergillus fumigatus versus Genus Aspergillus: Conservation, Adaptive Evolution and Specific Virulence Genes. Microorganisms, 9(10), 2014. https://doi.org/10.3390/microorganisms9102014