
3D Organoids: An Untapped Platform for Studying Host–Microbiome Interactions in Esophageal Cancers


Abstract
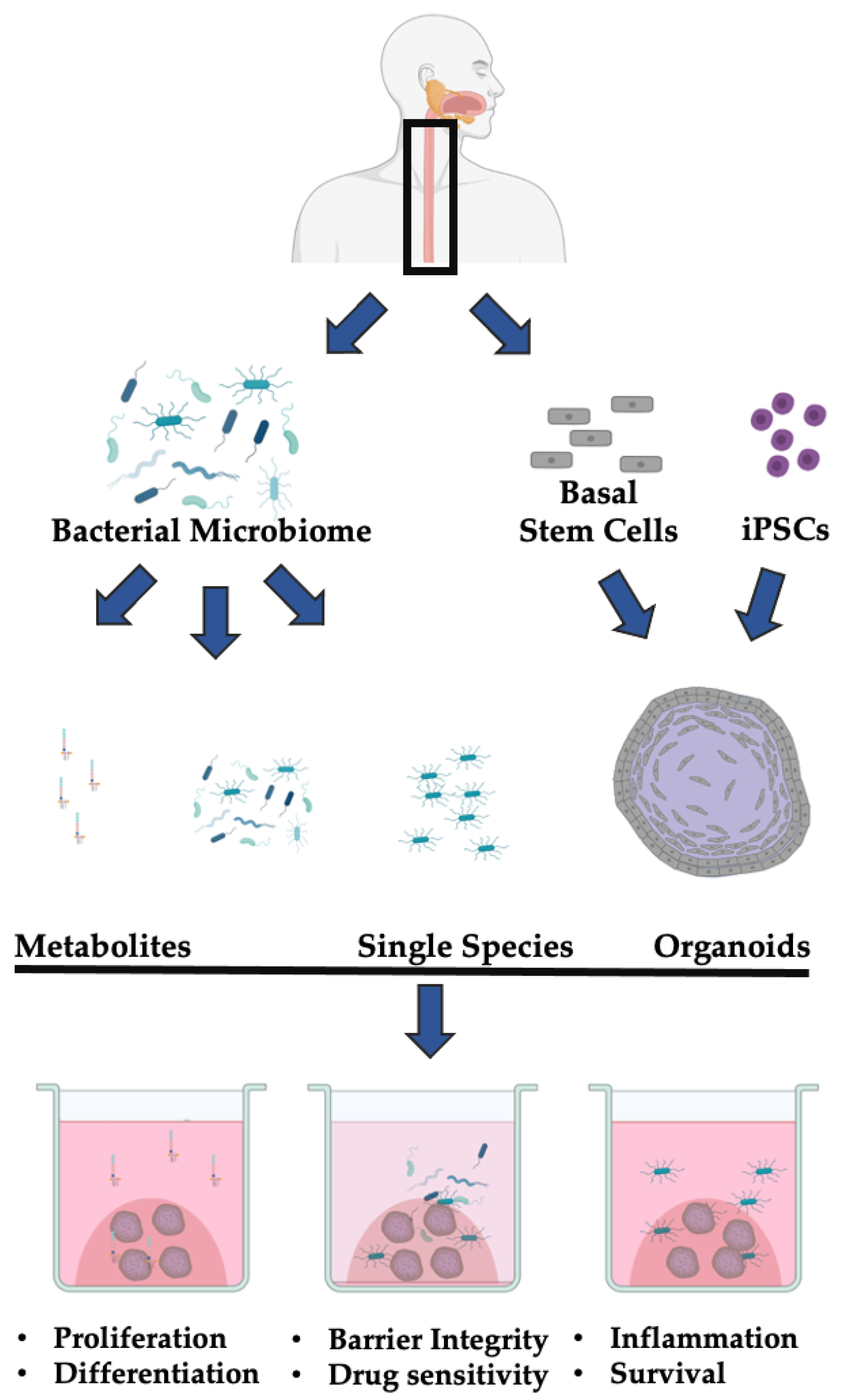
:1. Introduction
1.1. Esophageal Structure and Function
1.2. Esophageal Squamous Cell Carcinoma
1.3. Esophageal Adenocarcinoma
1.4. The Bacterial Microbiome in Esophageal Health and Disease
1.5. The 3D Esophageal Organoid System
2. Organoid and Microbiome Co-Culture Models of GI Cancer-Relevant Processes
2.1. Microbiome and Epithelial Cell Proliferation
2.2. Microbiome and Inflammation and Immunity
2.3. Microbiome and Mutagenesis
3. Discussion and Future Directions
3.1. Strengths and Weaknesses of the 3D Organoid-Microbiome Co-Culture Models
3.2. Utilizing 3D Organoid and Microbiome Co-Culture for the Study of Esophageal Health and Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The human microbiome and cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; Fitzgerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Uhlenhopp, D.J.; Then, E.O.; Sunkara, T.; Gaduputi, V. Epidemiology of esophageal cancer: Update in global trends, etiology and risk factors. Clin. J. Gastroenterol. 2020, 13, 1010–1021. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated With Alterations in the Microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumor microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef]
- Petrelli, F.; De Santi, G.; Rampulla, V.; Ghidini, A.; Mercurio, P.; Mariani, M.; Manara, M.; Rausa, E.; Lonati, V.; Viti, M.; et al. Human papillomavirus (HPV) types 16 and 18 infection and esophageal squamous cell carcinoma: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Yamamura, K.; Izumi, D.; Kandimalla, R.; Sonohara, F.; Baba, Y.; Yoshida, N.; Kodera, Y.; Baba, H.; Goel, A. Intratumoral Fusobacterium nucleatum levels predict therapeutic response to neoadjuvant chemotherapy in esophageal squamous cell carcinoma. Clin. Cancer Res. 2019, 25, 6170–6179. [Google Scholar] [CrossRef] [Green Version]
- Bruun, J.; Kryeziu, K.; Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Langerud, J.; Røsok, B.; Totland, M.Z.; Brunsell, T.H.; Pellinen, T.; et al. Patient-Derived Organoids from Multiple Colorectal Cancer Liver Metastases Reveal Moderate Intra-patient Pharmacotranscriptomic Heterogeneity. Clin. Cancer Res. 2020, 26, 4107–4119. [Google Scholar] [CrossRef] [Green Version]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.C.H.; Kranenburg, O.; Xiao, H.; Yu, J. Organoid models of gastrointestinal cancers in basic and translational research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 203–222. [Google Scholar] [CrossRef]
- Poletti, M.; Arnauts, K.; Ferrante, M.; Korcsmaros, T. Organoid-based Models to Study the Role of Host-microbiota Interactions in IBD. J. Crohn’s Colitis 2021, 15, 1222–1235. [Google Scholar] [CrossRef]
- Min, S.; Kim, S.; Cho, S.W. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp. Mol. Med. 2020, 52, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.D.; Orlando, R.C. Esophageal submucosal glands: Structure and function. Am. J. Gastroenterol. 1999, 94, 2818–2824. [Google Scholar] [CrossRef]
- Blevins, C.H.; Iyer, P.G.; Vela, M.F.; Katzka, D.A. The Esophageal Epithelial Barrier in Health and Disease. Clin. Gastroenterol. Hepatol. 2018, 16, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, Y.; Nakagawa, H.; Rustgi, A.K.; Que, J.; Wang, T.C. Stem cells and origins of cancer in the upper gastrointestinal tract. Stem Cell 2021, 28, 1343–1361. [Google Scholar] [CrossRef]
- Doupé, D.P.; Alcolea, M.P.; Roshan, A.; Zhang, G.; Klein, A.M.; Simons, B.D.; Jones, P.H. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 2012, 337, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef]
- Seery, J.P.; Watt, F.M. Asymmetric stem-cell divisions define the architecture of human oesophageal epithelium. Curr. Biol. 2000, 10, 1447–1450. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Nicholson, A.M.; Barr, H.; Harrison, L.A.; Wilson, G.D.; Burkert, J.; Jeffery, R.; Alison, M.R.; Looijenga, L.; Lin, W.R.; et al. Identification of lineage-uncommitted, long-lived, label-retaining cells in healthy human esophagus and stomach, and in metaplastic esophagus. Gastroenterology 2013, 144, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Mönkemüller, K.; Wex, T.; Kuester, D.; Fry, L.C.; Kandulski, A.; Kropf, S.; Roessner, A.; Malfertheiner, P. Role of tight junction proteins in gastroesophageal reflux disease. BMC Gastroenterol. 2012, 12. [Google Scholar] [CrossRef] [Green Version]
- Dignass, A.U.; Podolsky, D.K. Cytokine modulation of intestinal epithelial cell restitution: Central role of transforming growth factor beta. Gastroenterology 1993, 105, 1323–1332. [Google Scholar] [CrossRef]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Rustgi, A.K. Esophageal Cancers and Model Systems. Trans. Am. Clin. Climatol. Assoc. 2019, 130, 266–271. [Google Scholar]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Nakagawa, H.; Zukerberg, L.; Togawa, K.; Meltzer, S.J.; Nishihara, T.; Rustgi, A.K. Human cyclin D1 oncogene and esophageal squamous cell carcinoma. Cancer 1995, 76, 541–549. [Google Scholar] [CrossRef]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 508, 91–95. [Google Scholar] [CrossRef]
- Liu, K.; Jiang, M.; Lu, Y.; Chen, H.; Sun, J.; Wu, S.; Ku, W.Y.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; et al. Sox2 cooperates with inflammation-mediated stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 2013, 12, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamangar, F.; Nasrollahzadeh, D.; Safiri, S.; Sepanlou, S.G.; Fitzmaurice, C.; Ikuta, K.S.; Bisignano, C.; Islami, F.; Roshandel, G.; Lim, S.S.; et al. The global, regional, and national burden of oesophageal cancer and its attributable risk factors in 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 582–597. [Google Scholar] [CrossRef]
- Wang, L.D.; Zhou, F.Y.; Li, X.M.; Sun, L.D.; Song, X.; Jin, Y.; Li, J.M.; Kong, G.Q.; Qi, H.; Cui, J.; et al. Genome-wide association study of esophageal squamous cell carcinoma in chinese subjects identifies a susceptibility locus at PLCE1. Nat. Genet. 2010, 42, 759–765. [Google Scholar] [CrossRef]
- Menya, D.; Maina, S.K.; Kibosia, C.; Kigen, N.; Oduor, M.; Some, F.; Chumba, D.; Ayuo, P.; Middleton, D.R.S.; Osano, O.; et al. Dental fluorosis and oral health in the African Esophageal Cancer Corridor: Findings from the Kenya ESCCAPE case–control study and a pan-African perspective. Int. J. Cancer 2019, 145, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Yano, Y.; Etemadi, A.; Abnet, C.C. Microbiome and Cancers of the Esophagus: A Review. Microorganisms 2021, 9, 1764. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.A.C.; Lavery, D.; Wright, N.A.; Jansen, M. Barrett oesophagus: Lessons on its origins from the lesion itself. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 50–60. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Urbanska, A.M.; Hayakawa, Y.; Wang, H.; Au, A.S.; Luna, A.M.; Chang, W.; Jin, G.; Bhagat, G.; Abrams, J.A.; et al. Gastrin stimulates a cholecystokinin-2-receptor-expressing cardia progenitor cell and promotes progression of Barrett’s-like esophagus. Oncotarget 2017, 8, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Snider, E.J.; Freedberg, D.E.; Abrams, J.A. Potential Role of the Microbiome in Barrett’s Esophagus and Esophageal Adenocarcinoma. Dig. Dis. Sci. 2016, 61, 2217–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.; Lam, S.; Adam, M.; Gilroy, R.; Pallen, M.J. The oesophageal microbiome and cancer: Hope or hype? Trends Microbiol. 2021. [Google Scholar] [CrossRef]
- May, M.; Abrams, J.A. Emerging Insights into the Esophageal Microbiome. Curr. Treat. Options Gastroenterol. 2018, 16, 72–85. [Google Scholar] [CrossRef]
- Gagliardi, D.; Makihara, S.; Corsi, P.R.; De Toledo Viana, A.; Wiczer, M.V.F.S.; Nakakubo, S.; Mimica, L.M.J. Microbial flora of the normal esophagus. Dis. Esophagus 1998, 11, 248–250. [Google Scholar] [CrossRef]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef]
- Annavajhala, M.K.; May, M.; Compres, G.; Freedberg, D.E.; Graham, R.; Stump, S.; Que, J.; Korem, T.; Uhlemann, A.C.; Abrams, J.A. Relationship of the Esophageal Microbiome and Tissue Gene Expression and Links to the Oral Microbiome: A Randomized Clinical Trial. Clin. Transl. Gastroenterol. 2020, 11, e00235. [Google Scholar] [CrossRef]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Dong, L.; Zhao, J.; Wang, H.; Li, J.; Yu, A.; Chen, W.; Wei, W. Composition and consistence of the bacterial microbiome in upper, middle and lower esophagus before and after Lugol’s iodine staining in the esophagus cancer screening. Scand. J. Gastroenterol. 2020, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Gail, M.H.; Shi, J.; Klepac-Ceraj, V.; Paster, B.J.; Dye, B.A.; Wang, G.Q.; Wei, W.Q.; Fan, J.H.; Qiao, Y.L.; et al. Association between upper digestive tract microbiota and cancer-predisposing states in the esophagus and stomach. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Winckler, B.; Lu, M.; Cheng, H.; Yuan, Z.; Yang, Y.; Jin, L.; Ye, W. Oral microbiota and risk for esophageal squamous cell carcinoma in a high-risk area of China. PLoS ONE 2015, 10, e0143603. [Google Scholar] [CrossRef]
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Blackett, K.L.; Siddhi, S.S.; Cleary, S.; Steed, H.; Miller, M.H.; MacFarlane, S.; MacFarlane, G.T.; Dillon, J.F. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett’s and oesophageal carcinoma: Association or causality? Aliment. Pharmacol. Ther. 2013, 37, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.R.F.; Walker, A.W.; O’Donovan, M.; Parkhill, J.; Fitzgerald, R.C. A non-endoscopic device to sample the oesophageal microbiota: A case-control study. Lancet Gastroenterol. Hepatol. 2017, 2, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Liu, M.; Liu, Y.; Guo, C.; Zhou, Y.; Li, F.; Xu, R.; Liu, Z.; Deng, Q.; Li, X.; et al. Oral microbiome and risk of malignant esophageal lesions in a high-risk area of China: A nested case-control study. Chin. J. Cancer Res. 2020, 32, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Vogtmann, E.; Liu, A.; Qin, J.; Chen, W.; Abnet, C.C.; Wei, W. Microbial characterization of esophageal squamous cell carcinoma and gastric cardia adenocarcinoma from a high-risk region of China. Cancer 2019, 125, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Furrie, E.; Macfarlane, G.T.; Dillon, J.F. Microbial colonization of the upper gastrointestinal tract in patients with Barrett’s esophagus. Clin. Infect. Dis. 2007, 45, 29–38. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 2017, 77, 6777–6787. [Google Scholar] [CrossRef] [Green Version]
- Lopetuso, L.R.; Severgnini, M.; Pecere, S.; Ponziani, F.R.; Boskoski, I.; Larghi, A.; Quaranta, G.; Masucci, L.; Ianiro, G.; Camboni, T.; et al. Esophageal microbiome signature in patients with Barrett’s esophagus and esophageal adenocarcinoma. PLoS ONE 2020, 15, e0231789. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Baba, Y.; Ishimoto, T.; Tsutsuki, H.; Zhang, T.; Nomoto, D.; Okadome, K.; Yamamura, K.; Harada, K.; Eto, K.; et al. Fusobacterium nucleatum confers chemoresistance by modulating autophagy in oesophageal squamous cell carcinoma. Br. J. Cancer 2021, 124, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Lu, M.S.; Hsieh, C.C.; Chen, W.C. Porphyromonas gingivalis promotes tumor progression in esophageal squamous cell carcinoma. Cell. Oncol. 2021, 44, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, N.P.; Riordan, S.M.; Gorman, C.J.; Nielsen, S.; Russell, T.L.; Correa-Ospina, C.; Fernando, B.S.M.; Waters, S.A.; Castaño-Rodríguez, N.; Man, S.M.; et al. Multi-omics of the esophageal microenvironment identifies signatures associated with progression of Barrett’s esophagus. Genome Med. 2021, 13, 133. [Google Scholar] [CrossRef]
- Namin, B.M.; Dallal, M.M.S.; Daryani, N.E. The effect of Campylobacter concisus on expression of IL-18, TNF-α and p53 in barrett’s cell lines. Jundishapur J. Microbiol. 2015, 8. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, U.M.; Shimonosono, M.; Flashner, S.; Cruz-Acuña, R.; Gabre, J.T.; Nakagawa, H. Understanding the cellular origin and progression of esophageal cancer using esophageal organoids. Cancer Lett. 2021, 509, 39–52. [Google Scholar] [CrossRef]
- Kijima, T.; Nakagawa, H.; Shimonosono, M.; Chandramouleeswaran, P.M.; Hara, T.; Sahu, V.; Kasagi, Y.; Kikuchi, O.; Tanaka, K.; Giroux, V.; et al. Three-Dimensional Organoids Reveal Therapy Resistance of Esophageal and Oropharyngeal Squamous Cell Carcinoma Cells. Cmgh 2019, 7, 73–91. [Google Scholar] [CrossRef] [Green Version]
- Kasagi, Y.; Chandramouleeswaran, P.M.; Whelan, K.A.; Tanaka, K.; Giroux, V.; Sharma, M.; Wang, J.; Benitez, A.J.; DeMarshall, M.; Tobias, J.W.; et al. The Esophageal Organoid System Reveals Functional Interplay Between Notch and Cytokines in Reactive Epithelial Changes. Cmgh 2018, 5, 333–352. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Kasagi, Y.; Karakasheva, T.A.; Hara, T.; Aaron, B.; Shimonosono, M.; Kijima, T.; Giroux, V.; Bailey, D.; Wilkins, B.; et al. Modeling Epithelial Homeostasis and Reactive Epithelial Changes in Human and Murine Three-Dimensional Esophageal Organoids. Curr. Protoc. Stem Cell Biol. 2020, 52, e106. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Clevers, H.; Sato, T. Modeling Human Digestive Diseases With CRISPR-Cas9–Modified Organoids. Gastroenterology 2019, 156, 562–576. [Google Scholar] [CrossRef] [Green Version]
- DeWard, A.D.; Cramer, J.; Lagasse, E. Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep. 2014, 9, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroux, V.; Lento, A.A.; Islam, M.; Pitarresi, J.R.; Kharbanda, A.; Hamilton, K.E.; Whelan, K.A.; Long, A.; Rhoades, B.; Tang, Q.; et al. Long-lived keratin 15+ esophageal progenitor cells contribute to homeostasis and regeneration. J. Clin. Invest. 2017, 127, 2378–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Acuña, R.; Vunjak-Novakovic, G.; Burdick, J.A.; Rustgi, A.K. Emerging technologies provide insights on cancer extracellular matrix biology and therapeutics. iScience 2021, 24, 102475. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Kijima, T.; Shimonosono, M.; Maekawa, H.; Sahu, V.; Gabre, J.T.; Cruz-Acuña, R.; Giroux, V.; Sangwan, V.; Whelan, K.A.; et al. Generation and Characterization of Patient-Derived Head and Neck, Oral, and Esophageal Cancer Organoids. Curr. Protoc. Stem Cell Biol. 2020, 53, 1–27. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Y.; Jiang, M.; Huang, S.X.; Zhang, W.; Al Alam, D.; Danopoulos, S.; Mori, M.; Chen, Y.W.; Balasubramanian, R.; et al. 3D Modeling of Esophageal Development using Human PSC-Derived Basal Progenitors Reveals a Critical Role for Notch Signaling. Cell Stem Cell 2018, 23, 516–529.e5. [Google Scholar] [CrossRef] [Green Version]
- Trisno, S.L.; Philo, K.E.D.; McCracken, K.W.; Catá, E.M.; Ruiz-Torres, S.; Rankin, S.A.; Han, L.; Nasr, T.; Chaturvedi, P.; Rothenberg, M.E.; et al. Esophageal Organoids from Human Pluripotent Stem Cells Delineate Sox2 Functions during Esophageal Specification. Cell Stem Cell 2018, 23, 501–515.e7. [Google Scholar] [CrossRef] [Green Version]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Martinez-Silgado, A.; Akkerman, N.; Saftien, A.; Boot, C.; de Waal, A.; Beumer, J.; Dutta, D.; Heo, I.; et al. Intestinal organoid cocultures with microbes. Nat. Protoc. 2021, 16, 4633–4649. [Google Scholar] [CrossRef]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Clevers, H. Organoids and organs-on-chips: Insights into human gut-microbe interactions. Cell Host Microbe 2021, 29, 867–878. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [Green Version]
- McCracken, K.W.; Catá, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M., Jr.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 Plays a Functional Role in Helicobacter pylori-induced Epithelial Cell Proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef]
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like Receptor 4 Is Expressed on Intestinal Stem Cells and Regulates Their Proliferation and Apoptosis via the p53 Up-regulated Modulator of Apoptosis. J. Biol. Chem. 2012, 287, 37296–37308. [Google Scholar] [CrossRef] [Green Version]
- Naito, T.; Mulet, C.; De Castro, C.; Molinaro, A.; Saffarian, A.; Nigro, G.; Bérard, M.; Clerc, M.; Pedersen, A.B.; Sansonetti, P.J.; et al. Lipopolysaccharide from crypt-specific core microbiota modulates the colonic epithelial proliferation-to-differentiation balance. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bartfeld, S.; Bayram, T.; Van De Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136.e6. [Google Scholar] [CrossRef] [Green Version]
- Bartfeld, S.; Hess, S.; Bauer, B.; Machuy, N.; Ogilvie, L.A.; Schuchhardt, J.; Meyer, T.F. High-throughput and single-cell imaging of NF-κB oscillations using monoclonal cell lines. BMC Cell Biol. 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Ferrero, R.L. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol. Immunol. 2005, 42, 879–885. [Google Scholar] [CrossRef]
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.; Yu, Q. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018, 25, 1657–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, D.R.; Huang, S.; Nagy, M.S.; Yadagiri, V.K.; Fields, C.; Mukherjee, D.; Bons, B.; Dedhia, P.H.; Chin, A.M.; Tsai, Y.H.; et al. Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Broda, T.; Chang, J.; Hawkins, J.A.; Sundaram, N.; Wroblewski, L.E.; Peek, R.M.; Wang, J.; Helmrath, M.; et al. Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection. PLoS Pathog. 2019, 15, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Sebrell, T.A.; Hashimi, M.; Sidar, B.; Wilkinson, R.A.; Kirpotina, L.; Quinn, M.T.; Malkoç, Z.; Taylor, P.J.; Wilking, J.N.; Bimczok, D. A Novel Gastric Spheroid Co-culture Model Reveals Chemokine-Dependent Recruitment of Human Dendritic Cells to the Gastric Epithelium. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 157–171.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks + E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Luca, F.; Kupfer, S.S.; Knights, D.; Khoruts, A.; Blekhman, R. Functional Genomics of Host-Microbiome Interactions in Humans. Trends Genet. 2018, 34, 30–40. [Google Scholar] [CrossRef]
- Goto, Y. Commensal bacteria prevent pathogenic bacterial infection by inducing of activation of host immune system. Nippon Saikingaku Zasshi 2020, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, S.; Ma, Z.; Liang, S.; Shan, T.; Zhang, M.; Zhu, X.; Zhang, P.; Liu, G.; Zhou, F.; et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect. Agent. Cancer 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Liu, Y.; Duan, X.; Liu, K.; Mohammed, M.; Gu, Z.; Ren, J.; Yakoumatos, L.; Yuan, X.; Lu, L.; et al. Porphyromonas gingivalis infection exacerbates oesophageal cancer and promotes resistance to neoadjuvant chemotherapy. Br. J. Cancer 2021, 125, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Rajendra, S.; Pavey, D.; McKay, O.; Merrett, N.; Gautam, S.D. Human papillomavirus infection in esophageal squamous cell carcinoma and esophageal adenocarcinoma: A concise review. Ann. N. Y. Acad. Sci. 2020, 1482, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hisamatsu, K.; Suzui, N.; Hara, A.; Tomita, H.; Miyazaki, T. A Review of HPV-Related Head and Neck Cancer. J. Clin. Med. 2018, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.Y.; Xu, L.Y.; Chen, M.H.; Shen, J.; Cai, W.J.; Zeng, Y. Progressive transformation of immortalized esophageal epithelial cells. World J. Gastroenterol. 2002, 8, 976–981. [Google Scholar] [CrossRef]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lõhmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral mucosal organoids as a potential platform for personalized cancer therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef]
- Ma, T.; Ru, J.; Xue, J.; Schulz, S.; Mirzaei, M.K.; Janssen, K.-P.; Quante, M.; Deng, L. Differences in Gut Virome Related to Barrett Esophagus and Esophageal Adenocarcinoma. Microorganisms 2021, 9, 1701. [Google Scholar] [CrossRef]
- Ajayi, T.A.; Cantrell, S.; Spann, A.; Garman, K.S. Barrett’s esophagus and esophageal cancer: Links to microbes and the microbiome. PLoS Pathog. 2018, 14, e1007384. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Kelly, L.A.; Kreft, R.E.; Barlek, M.; Omstead, A.N.; Matsui, D.; Boyd, N.H.; Gazarik, K.E.; Heit, M.I.; Nistico, L.; et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer 2016. [Google Scholar] [CrossRef] [Green Version]
- Bonavina, L.; Incarbone, R.; Reitano, M.; Tortorano, A.; Viviani, M.; Peracchia, A. Candida colonization in patients with esophageal disease: A prospective clinical study. Dis. Esophagus 2003, 16, 70–72. [Google Scholar] [CrossRef]
- Rosa, D.D.; Pasqualotto, A.C.; Denning, D.W. Chronic mucocutaneous candidiasis and oesophageal cancer. Med. Mycol. 2008, 46, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delsing, C.E.; Bleeker-Rovers, C.P.; van de Veerdonk, F.L.; Tol, J.; van der Meer, J.W.M.; Kullberg, B.J.; Netea, M.G. Association of esophageal candidiasis and squamous cell carcinoma. Med. Mycol. Case Rep. 2012, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.-C.; Sun, T.-T.; Wang, Y.-Y.; Anderson, L.M.; Armstrong, D.; Good, R.A. Enhancement of formation of the esophageal carcinogen benzylmethylnitrosamine from its precursors by Candida albicans. Proc. Natl. Acad. Sci. USA 1981, 78, 1878–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittamo, J.; Siikala, E.; Kaihovaara, P.; Salaspuro, M.; Rautemaa, R. Chronic candidosis and oral cancer in APECED-patients: Production of carcinogenic acetaldehyde from glucose and ethanol by Candida albicans. Int. J. Cancer 2009, 124, 754–756. [Google Scholar] [CrossRef]


{kind=link}
| Tissue | Microbe | Classification | Product | Model | Host | Cancer-Associated Phenotype | Reference |
|---|---|---|---|---|---|---|---|
| Gastric | H. pylori | Pathogenic | Whole bacteria | Luminal microinjection | Human | Increased PD-L1 expression, increased survival | [77] |
| Increased inflammatory cytokine production (CXCL2, CXCL16, CXCL17, and CCL20), DC recruitment | [78] | ||||||
| Increased proliferation through c-Met signaling | [79] | ||||||
| Increased inflammatory cytokine production through the NF-κB pathway | [80] | ||||||
| Mouse | Increased proliferation through β-catenin signaling, mislocalization of Claudin-7 | [81] | |||||
| Human; Mouse | Increased CD44-dependent proliferation and EMT | [82] | |||||
| Intestinal | pks + E. coli | Pathogenic | Whole bacteria | Luminal microinjection | Human | Increased DNA damage and mutational burden | [83] |
| Mouse | Increased proliferation, decreased differentiation, increased chromosomal alterations, increased DNA mutational burden | [84] | |||||
| E. coli | Commensal | Whole bacteria | Luminal microinjection | Human | Increased proliferation (transient), enhanced barrier integrity through IL-6 and IL-8 signaling | [85] | |
| LPS | Supplemented into media | Mouse | Decreased proliferation, increased apoptosis through TLR4 signaling | [86] | |||
| Acinetobacter, Stenotrophomonas, and Delftia genera | Commensal | LPS | Supplemented into media | Mouse | Decreased proliferation, increased necroptosis, increased differentiation through TLR4 signaling | [87] | |
| L. reuteri D8 | Commensal | Whole bacteria, indole-3-aldehyde | Supplemented into media | Mouse | Increased proliferation, enhanced barrier integrity through IL-22 signaling | [88] | |
| Common commensal metabolites | Commensal | Gallic acid | Supplemented into media | Mouse | Increased WNT signaling, Increased proliferation, decreased differentiation in mutant p53 epithelial cells | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flashner, S.; Yan, K.S.; Nakagawa, H. 3D Organoids: An Untapped Platform for Studying Host–Microbiome Interactions in Esophageal Cancers. Microorganisms 2021, 9, 2182. https://doi.org/10.3390/microorganisms9112182
Flashner S, Yan KS, Nakagawa H. 3D Organoids: An Untapped Platform for Studying Host–Microbiome Interactions in Esophageal Cancers. Microorganisms. 2021; 9(11):2182. https://doi.org/10.3390/microorganisms9112182
Chicago/Turabian StyleFlashner, Samuel, Kelley S. Yan, and Hiroshi Nakagawa. 2021. "3D Organoids: An Untapped Platform for Studying Host–Microbiome Interactions in Esophageal Cancers" Microorganisms 9, no. 11: 2182. https://doi.org/10.3390/microorganisms9112182
APA StyleFlashner, S., Yan, K. S., & Nakagawa, H. (2021). 3D Organoids: An Untapped Platform for Studying Host–Microbiome Interactions in Esophageal Cancers. Microorganisms, 9(11), 2182. https://doi.org/10.3390/microorganisms9112182