
Metagenomic Investigation of a Low Diversity, High Salinity Offshore Oil Reservoir

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of Formation Water
2.2. Water Analysis
2.3. DNA Extraction, 16S rRNA Gene Sequencing and Analysis
2.4. Metagenomics
2.4.1. Metagenome Sequencing
2.4.2. Processing of Metagenome-Assembled Genomes (MAGs)
2.4.3. Metabolic Predictions
2.4.4. Phylogeny
3. Results and Discussion
3.1. Formation Water Chemistry
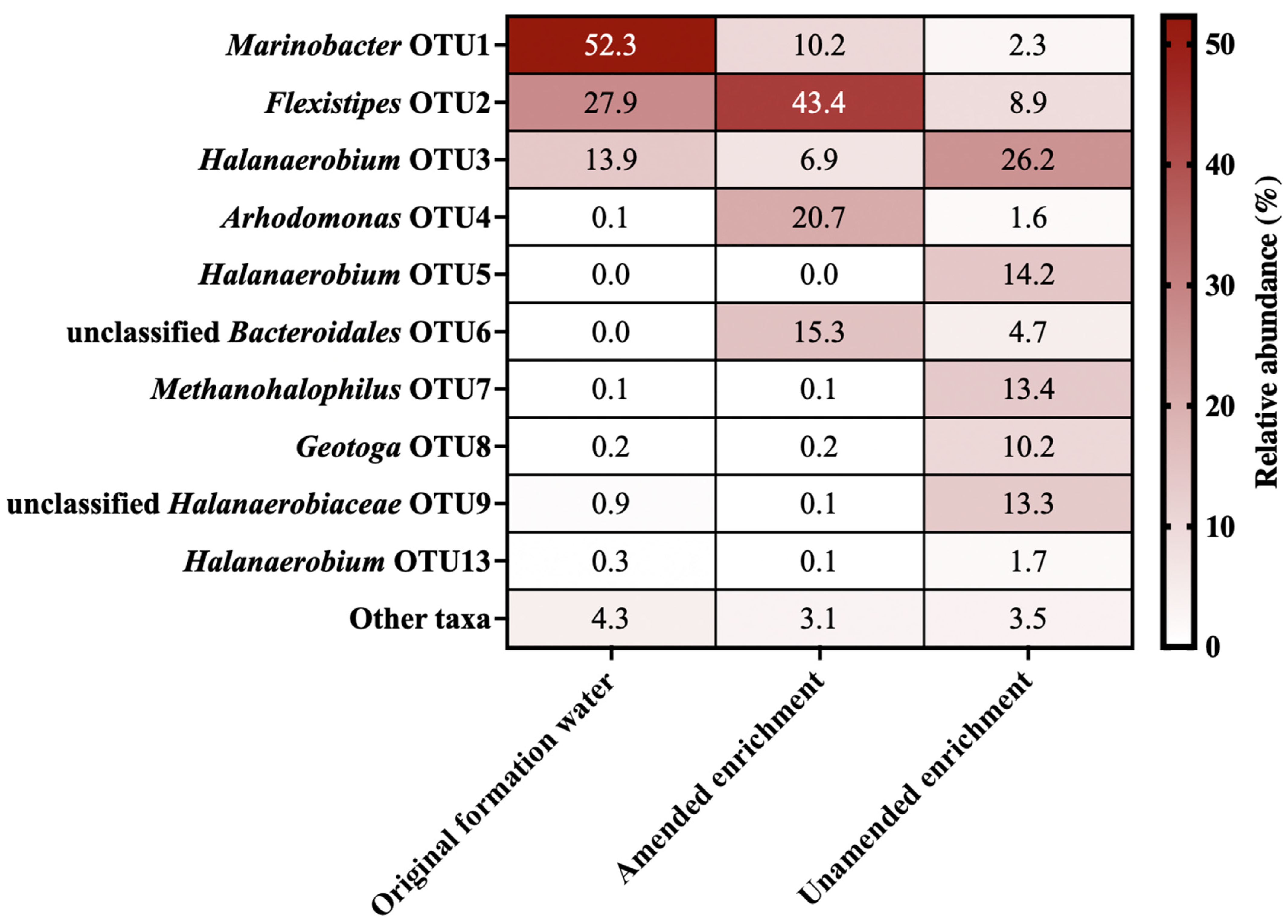
3.2. Microbial Community Composition
3.3. Metagenome Assembled Genomes and Key Metabolisms from a High Salinity Offshore Oil Reservoir
3.3.1. Carbon Metabolism
Fermentation
Hydrocarbon Biodegradation
Methanogenesis
3.3.2. Sulfur Metabolism
SOX System
Thiosulfate Reduction
3.3.3. Nitrogen Metabolism
Dissimilatory Nitrate Reduction to Ammonia (DNRA)
Denitrification
3.3.4. Nitrogen and Sulfur Metabolism in the Context of Souring and Souring Control
3.4. Novel Lineages
3.4.1. Reconstructed Genome of BM520
3.4.2. Reconstructed Genome of QPJE01
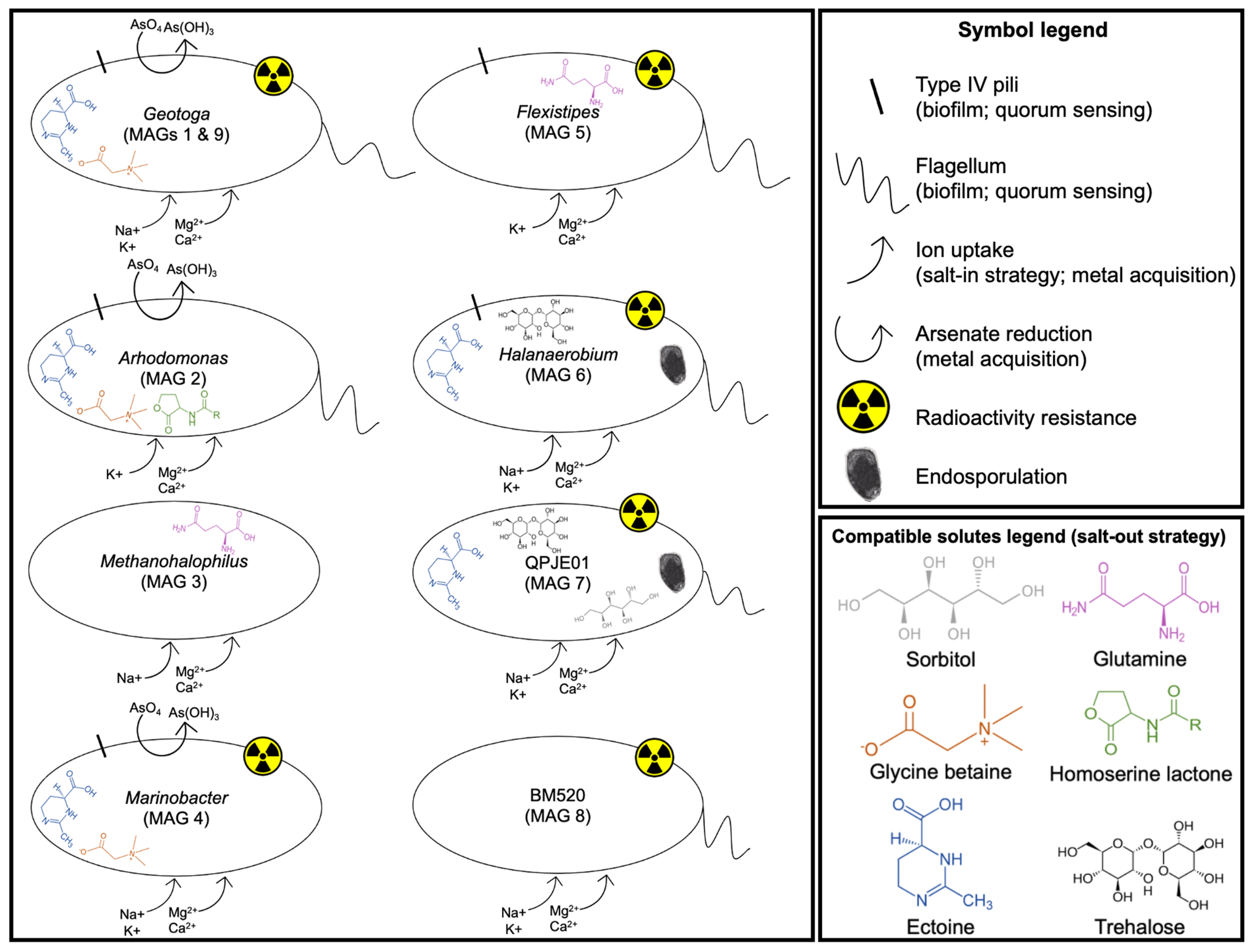
3.5. Adaptation to Extreme Environmental Conditions
3.5.1. Salt Adaptation
Salt-In Strategy
Salt-Out Strategy
3.5.2. Radiation Adaptation
3.5.3. Metal Acquisition
3.5.4. Endosporulation
3.5.5. Quorum Sensing and Biofilm Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohli, I.; Joshi, N.C.; Mohapatra, S.; Varma, A. Extremophile—An adaptive strategy for extreme conditions and applications. Curr. Genom. 2020, 21, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Merino, N.; Aronson, H.S.; Bojanova, D.P.; Feyhl-Buska, J.; Wong, M.L.; Zhang, S.; Giovannelli, D. Living at the extremes: Extremophiles and the limits of life in a planetary context. Front. Microbiol. 2019, 10, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Rampelotto, P.H. Extremophiles and extreme environments. Life 2013, 3, 482–485. [Google Scholar] [CrossRef]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef]
- Pannekens, M.; Kroll, L.; Müller, H.; Mbow, F.T.; Meckenstock, R.U. Oil reservoirs, an exceptional habitat for microorganisms. New Biotechnol. 2019, 49, 1–9. [Google Scholar] [CrossRef]
- US EPA. Radioactive Waste Material from Oil and Gas Drilling. Available online: https://www.epa.gov/radtown/radioactive-waste-material-oil-and-gas-drilling (accessed on 20 May 2021).
- Youssef, N.; Elshahed, M.S.; McInerney, M.J. Microbial processes in oil fields: Culprits, problems, and opportunities. Adv. Appl. Microbiol. 2009, 66, 141–251. [Google Scholar]
- Sierra-Garcia, I.N.; de Oliveira, V.M. Microbial Hydrocarbon Degradation: Efforts to Understand Biodegradation in Petroleum Reservoirs; IntechOpen: London, UK, 2013. [Google Scholar]
- Guan, J.; Zhang, B.-L.; Mbadinga, S.M.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z. Functional genes (Dsr) approach reveals similar sulphidogenic prokaryotes diversity but different structure in saline waters from corroding high temperature petroleum reservoirs. Appl. Microbiol. Biotechnol. 2014, 98, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Oliveira, D.A.F.; Holsten, L.; Steinhauer, K.; de Rezende, J.R. Long-term biocide efficacy and its effect on a souring microbial community. Appl. Environ. Microbiol. 2021, 87, AEM0084221. [Google Scholar] [CrossRef]
- An, B.A.; Shen, Y.; Voordouw, G. Control of sulfide production in high salinity Bakken shale oil reservoirs by halophilic bacteria reducing nitrate to nitrite. Front. Microbiol. 2017, 8, 1164. [Google Scholar] [CrossRef] [Green Version]
- Veshareh, M.J.; Nick, H. Learnings from reservoir souring treatment by nitrate injection in the Halfdan oil field. In Proceedings of the 80th EAGE Conference and Exhibition 2018, Copenhagen, Denmark, 11–14 June 2018; pp. 1–5. [Google Scholar]
- Payler, S.J.; Biddle, J.F.; Lollar, B.S.; Fox-Powell, M.G.; Edwards, T.; Ngwenya, B.T.; Paling, S.M.; Cockell, C.S. An ionic limit to life in the deep subsurface. Front. Microbiol. 2019, 10, 426–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Lanzen, A.; Davenport, R.J.; Turnbaugh, P.J. Removing noise from pyrosequenced amplicons. BMC Bioinform. 2011, 12, 38. [Google Scholar] [CrossRef]
- De Rezende, J.R.; Oldenburg, T.B.P.; Korin, T.; Richardson, W.D.L.; Fustic, M.; Aitken, C.M.; Bowler, B.F.J.; Sherry, A.; Grigoryan, A.; Voordouw, G.; et al. Anaerobic microbial communities and their potential for bioenergy production in heavily biodegraded petroleum reservoirs. Environ. Microbiol. 2020, 22, 3049–3065. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16s RRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JGI BBDuk Guide. Available online: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/ (accessed on 31 October 2020).
- FastQC—A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 April 2021).
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an eficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, 1165. [Google Scholar] [CrossRef] [Green Version]
- Bowtie 2: Fast and Sensitive Read Alignment. Available online: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml (accessed on 26 September 2020).
- Genome Research Limited Samtools. Available online: http://www.htslib.org/ (accessed on 28 April 2021).
- An analysis and Visualization Platform For ’Omics Data. Available online: https://merenlab.org/software/anvio/ (accessed on 28 April 2021).
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Daly, R.A.; Borton, M.A.; Wilkins, M.J.; Hoyt, D.W.; Kountz, D.J.; Wolfe, R.A.; Welch, S.A.; Marcus, D.N.; Trexler, R.V.; MacRae, J.D.; et al. Microbial metabolisms in a 2.5-km-deep ecosystem created by hydraulic fracturing in shales. Nat. Microbiol. 2016, 1, 16146. [Google Scholar] [CrossRef] [PubMed]
- Ryabova, A.; Kozlova, O.; Kadirov, A.; Ananeva, A.; Gusev, O.; Shagimardanova, E. DetR DB: A database of ionizing radiation resistance determinants. Genes 2020, 11, 1477. [Google Scholar] [CrossRef]
- Jones, A.A.; Pilloni, G.; Claypool, J.T.; Paiva, A.R.; Summers, Z.M. Evidence of sporulation capability of the ubiquitous oil reservoir microbe Halanaerobium congolense. Geomicrobiol. J. 2020, 38, 283–293. [Google Scholar] [CrossRef]
- Protein BLAST: Search Protein Databases Using a Protein Query. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome (accessed on 28 April 2021).
- Huson, D.H.; Rupp, R.; Berry, V.; Gambette, P.; Paul, C. Computing galled networks from real data. Bioinformatics 2009, 25, i85–i93. [Google Scholar] [CrossRef] [PubMed]
- Head, I.M.; Gray, N.D.; Larter, S.R. Life in the slow lane; biogeochemistry of biodegraded petroleum containing reservoirs and implications for energy recovery and carbon management. Front. Microbiol. 2014, 5, 566. [Google Scholar] [CrossRef] [Green Version]
- Grassia, G.S.; McLean, K.M.; Glénat, P.; Bauld, J.; Sheehy, A.J. A systematic survey for thermophilic fermentative bacteria and archaea in high temperature petroleum reservoirs. FEMS Microbiol. Ecol. 1996, 21, 47–58. [Google Scholar] [CrossRef]
- Röling, W.F.M.; Head, I.M.; Larter, S.R. The microbiology of hydrocarbon degradation in subsurface petroleum reservoirs: Perspectives and prospects. Res. Microbiol. 2003, 154, 321–328. [Google Scholar] [CrossRef]
- Gray, N.D.; Sherry, A.; Larter, S.R.; Erdmann, M.; Leyris, J.; Liengen, T.; Beeder, J.; Head, I.M. Biogenic methane production in formation waters from a large gas field in the North Sea. Extremophiles 2009, 13, 511–519. [Google Scholar] [CrossRef]
- Waldron, P.J.; Petsch, S.T.; Martini, A.M.; Nüsslein, K. Salinity constraints on subsurface archaeal diversity and methanogenesis in sedimentary rock rich in organic matter. Appl. Environ. Microbiol. 2007, 73, 4171–4179. [Google Scholar] [CrossRef] [Green Version]
- Binmerdhah, A.; Yassin, A. Solubility of common oil field scales of injection water and high–barium concentration and high–salinity formation water. J. Teknol. 2009, 50, 67–77. [Google Scholar]
- Gamal, H.; Al-Afnan, S.; Elkatatny, S.; Bahgat, M. Barium sulfate scale removal at low-temperature. Geofluids 2021, 2021, 5527818. [Google Scholar] [CrossRef]
- Mahmoud, M.; Elkatatny, S.; Abdelgawad, K.Z. Using high- and low-salinity seawater injection to maintain the oil reservoir pressure without damage. J. Pet. Explor. Prod. Technol. 2017, 7, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Borin, S.; Crotti, E.; Mapelli, F.; Tamagnini, I.; Corselli, C.; Daffonchio, D. DNA is preserved and maintains transforming potential after contact with brines of the deep anoxic hypersaline lakes of the Eastern Mediterranean sea. Saline Syst. 2008, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenova, E.M.; Grouzdev, D.S.; Tourova, T.P.; Nazina, T.N. Physiology and genomic characteristics of Geotoga petraea, a bacterium isolated from a low-temperature petroleum reservoir (Russia). Microbiology 2019, 88, 662–670. [Google Scholar] [CrossRef]
- Adkins, J.P.; Madigan, M.T.; Mandelco, L.; Woese, C.R.; Tanner, R.S. Arhodomonas aquaeolei gen. nov., sp. nov., an aerobic, halophilic bacterium isolated from a subterranean brine. Int. J. Syst. Bacteriol. 1993, 43, 514–520. [Google Scholar] [CrossRef] [Green Version]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-led, integrated, reproducible multi-omics with Anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Kaji, M.; Taniguchi, Y.; Matsushita, O.; Katayama, S.; Miyata, S.; Morita, S.; Okabe, A. The HydA gene encoding the H(2)-evolving hydrogenase of Clostridium perfringens: Molecular characterization and expression of the Gene. FEMS Microbiol. Lett. 1999, 181, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Boock, J.T.; Freedman, A.J.E.; Tompsett, G.A.; Muse, S.K.; Allen, A.J.; Jackson, L.A.; Castro-Dominguez, B.; Timko, M.T.; Prather, K.L.J.; Thompson, J.R. Engineered microbial biofuel production and recovery under supercritical carbon dioxide. Nat. Commun. 2019, 10, 587. [Google Scholar] [CrossRef] [Green Version]
- Ravot, G.; Magot, M.; Ollivier, B.; Patel, B.K.C.; Ageron, E.; Grimont, P.A.D.; Thomas, P.; Garcia, J.-L. Halanaerobium congolense sp. nov., an anaerobic, moderately halophilic, thiosulfate- and sulfur-reducing bacterium from an African oil field. FEMS Microbiol. Lett. 1997, 147, 81–88. [Google Scholar] [CrossRef]
- Chakraborty, A.; Ruff, S.E.; Dong, X.; Ellefson, E.D.; Li, C.; Brooks, J.M.; McBee, J.; Bernard, B.B.; Hubert, C.R.J. Hydrocarbon seepage in the deep seabed links subsurface and seafloor biospheres. Proc. Natl. Acad. Sci. USA 2020, 117, 11029–11037. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, Y.; Jeon, C.O. Biodegradation of naphthalene, BTEX, and aliphatic hydrocarbons by Paraburkholderia aromaticivorans BN5 isolated from petroleum-contaminated soil. Sci. Rep. 2019, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Bonfá, M.R.L.; Grossman, M.J.; Piubeli, F.; Mellado, E.; Durrant, L.R. Phenol degradation by halophilic bacteria isolated from hypersaline environments. Biodegradation 2013, 24, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Fathepure, B.Z. Recent studies in microbial degradation of petroleum hydrocarbons in hypersaline environments. Front. Microbiol. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, N.; Chandran, P. Microbial degradation of petroleum hydrocarbon contaminants: An overview. Biotechnol. Res. Int. 2010, 2011, 941810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Nguyi, D.K.; Vinu, M.; Blom, J.; Alam, I.; Guillot, S.; Ferry, J.S.; Stingl, U. Comparative genomics of the genus Methanohalophilus, including a newly isolated strain from Kebrit deep in the Red Sea. Front. Microbiol. 2019, 10, 839. [Google Scholar] [CrossRef]
- Grabarczyk, D.B.; Berks, B.C. Intermediates in the SOX sulfur oxidation pathway are bound to a sulfane conjugate of the carrier protein SoxYZ. PLoS ONE 2017, 12, e0173395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Voelker, J.; Keskin, C.; Xu, Z.; Hu, B.; Jia, C. A flow assurance study on elemental sulfur deposition in sour gas wells. In Proceedings of the SPE Annual Technical Conference and Exhibition, Denver, CO, USA, 30 October–2 November 2011. [Google Scholar]
- Anantharaman, K.; Hausmann, B.; Jungbluth, S.P.; Kantor, R.S.; Lavy, A.; Warren, L.A.; Rappé, M.S.; Pester, M.; Loy, A.; Thomas, B.C.; et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018, 12, 1715–1728. [Google Scholar] [CrossRef] [Green Version]
- Slobodkin, A.; Slobodkina, G.; Allioux, M.; Alain, K.; Jebbar, M.; Shadrin, V.; Kublanov, I.; Toshchakov, S.; Bonch-Osmolovskaya, E. Genomic insights into the carbon and energy metabolism of a thermophilic deep-sea bacterium Deferribacter autotrophicus revealed new metabolic traits in the phylum Deferribacteres. Genes 2019, 10, 849. [Google Scholar] [CrossRef] [Green Version]
- Coelho, C.; Romão, M.J. Structural and mechanistic insights on nitrate reductases. Protein Sci. 2015, 24, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Bødtker, G.; Lysnes, K.; Torsvik, T.; Bjørnestad, E.Ø.; Sunde, E. Microbial analysis of backflowed injection water from a nitrate-treated North Sea oil reservoir. J. Ind. Microbiol. Biotechnol. 2009, 36, 439–450. [Google Scholar] [CrossRef]
- Evans, M.V.; Panescu, J.; Hanson, A.J.; Welch, S.A.; Sheets, J.M.; Nastasi, N.; Daly, R.A.; Cole, D.R.; Darrah, T.H.; Wilkins, M.J.; et al. Members of Marinobacter and Arcobacter influence system biogeochemistry during early production of hydraulically fractured natural gas wells in the Appalachian Basin. Front. Microbiol. 2018, 9, 2646. [Google Scholar] [CrossRef]
- Carlson, H.; Hubert, C. Mechanisms and monitoring of oil reservoir souring control by nitrate or perchlorate injection. In Microbial Communities Utilizing Hydrocarbons and Lipds: Members, Metagenomics and Ecophysiology; Springer International Publishing: New York, NY, USA, 2019; pp. 1–25. [Google Scholar]
- Suri, N.; Voordouw, J.; Voordouw, G. The effectiveness of nitrate-mediated control of the oil field sulfur cycle depends on the toluene content of the oil. Front. Microbiol. 2017, 8, 956. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.D.; O’Farrell, C.; Toth, C.R.A.; Montoya, O.; Gieg, L.M.; Kwon, T.-H.; Yoon, S. Microbial community analyses of produced waters from high-temperature oil reservoirs reveal unexpected similarity between geographically distant oil reservoirs. Microb. Biotechnol. 2018, 11, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Y.; Gao, C.-X.; Mbadinga, S.M.; Zhou, L.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z. Characterization of an alkane-degrading methanogenic enrichment culture from production water of an oil reservoir after 274 days of incubation. Int. Biodeterior. Biodegrad. 2011, 65, 444–450. [Google Scholar] [CrossRef]
- Purwasena, I.A.; Sugai, Y.; Sasaki, K. The utilization of natural reservoir brine in an enrichment culture medium: An alternative approach for isolation of anaerobic bacteria from an oil reservoir. Pet. Sci. 2014, 32, 783–789. [Google Scholar] [CrossRef]
- Shelton, J.L.; Akob, D.M.; McIntosh, J.C.; Fierer, N.; Spear, J.R.; Warwick, P.D.; McCray, J.E. Environmental drivers of differences in microbial community structure in crude oil reservoirs across a methanogenic gradient. Front. Microbiol. 2016, 7, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gales, G.; Tsesmetzis, N.; Neria, I.; Alazard, D.; Coulon, S.; Lomans, B.P.; Morin, D.; Ollivier, B.; Borgomano, J.; Joulian, C. Preservation of ancestral cretaceous microflora recovered from a hypersaline oil reservoir. Sci. Rep. 2016, 6, 22960. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.-L.; Hanada, S.; Ohashi, A.; Harada, H.; Kamagata, Y.; Sekiguchi, Y. Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl. Environ. Microbiol. 2008, 74, 2051–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, N.; Eom, T.; Gupta, S.K.; Jeong, S.-Y.; Jeong, D.-Y.; Kim, Y.S.; Lee, J.-H.; Sadowsky, M.J.; Unno, T. Genes and gut bacteria involved in luminal butyrate reduction caused by diet and loperamide. Genes 2017, 8, 350. [Google Scholar] [CrossRef] [Green Version]
- Booker, A.E.; Borton, M.A.; Daly, R.A.; Welch, S.A.; Nicora, C.D.; Hoyt, D.W.; Wilson, T.; Purvine, S.O.; Wolfe, R.A.; Sharma, S.; et al. Sulfide generation by dominant Halanaerobium microorganisms in hydraulically fractured shales. MSphere 2017, 2, e00257-17. [Google Scholar] [CrossRef] [Green Version]
- Mouser, P.J.; Borton, M.; Darrah, T.H.; Hartsock, A.; Wrighton, K.C. Hydraulic fracturing offers view of microbial life in the deep terrestrial subsurface. FEMS Microbiol. Ecol. 2016, 92, fiw166. [Google Scholar] [CrossRef]
- Lipus, D.; Vikram, A.; Ross, D.; Bain, D.; Gulliver, D.; Hammack, R.; Bibby, K. Predominance and metabolic potential of Halanaerobium spp. in produced water from hydraulically fractured Marcellus shale wells. Appl. Environ. Microbiol. 2017, 83, e02659-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, A. Bioenergetic aspects of halophilism. Microbiol. Mol. Biol. Rev. 1999, 63, 334–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef] [PubMed]
- Kivistö, A.; Larjo, A.; Ciranna, A.; Santala, V.; Roos, C.; Karp, M. Genome sequence of Halanaerobium saccharolyticum subsp. saccharolyticum strain DSM 6643T, a halophilic hydrogen-producing bacterium. Genome Announc. 2013, 1, e00187-13. [Google Scholar] [PubMed] [Green Version]
- Ali, M.M.; Zhao, H.; Li, Z.; Maglas, N.N. Concentrations of TENORMs in the petroleum industry and their environmental and health effects. RSC Adv. 2019, 9, 39201–39229. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Aravind, L.; Wolf, Y.I.; Tatusov, R.L.; Minton, K.W.; Koonin, E.V.; Daly, M.J. Genome of the extremely radiation-resistant bacterium Deinococcus radiodurans viewed from the perspective of comparative genomics. Microbiol. Mol. Biol. Rev. 2001, 65, 44–79. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.I.; Cartwright, J.L.; Harashima, H.; Kamiya, H.; McLennan, A.G. Characterization of a Nudix hydrolase from Deinococcus radiodurans with a marked specificity for (deoxy)ribonucleoside 5′-diphosphates. BMC Biochem. 2004, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Dulermo, R.; Onodera, T.; Coste, G.; Passot, F.; Dutertre, M.; Porteron, M.; Confalonieri, F.; Sommer, S.; Pasternak, C. Identification of new genes contributing to the extreme radioresistance of Deinococcus radiodurans using a Tn5-based transposon mutant library. PLoS ONE 2015, 10, e0124358. [Google Scholar] [CrossRef] [Green Version]
- Wakeman, C.A.; Goodson, J.R.; Zacharia, V.M.; Winkler, W.C. Assessment of the requirements for magnesium transporters in Bacillus subtilis. J. Bacteriol. 2014, 196, 1206–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ruiz, F.; Jroundi, F.; Paytan, A.; Guerra-Tschuschke, I.; del Mar Abad, M.; González-Muñoz, M.T. Barium bioaccumulation by bacterial biofilms and implications for Ba cycling and use of Ba proxies. Nat. Commun. 2018, 9, 1619. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Muñoz, M.T.; Martinez-Ruiz, F.; Morcillo, F.; Martin-Ramos, J.D.; Paytan, A. Precipitation of barite by marine bacteria: A possible mechanism for marine barite formation. Geology 2012, 40, 675–678. [Google Scholar] [CrossRef]
- Wackett, L.P.; Dodge, A.G.; Ellis, L.B.M. Microbial genomics and the periodic table. Appl. Environ. Microbiol. 2004, 70, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.D.; Galera-Laporta, L.; Bialecka-Fornal, M.; Moon, E.C.; Shen, Z.; Briggs, S.P.; Garcia-Ojalvo, J.; Süel, G.M. Magnesium flux modulates ribosomes to increase bacterial survival. Cell 2019, 177, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, S.; Phung, L.T. Genes and enzymes involved in bacterial oxidation and reduction of inorganic arsenic. Appl. Environ. Microbiol. 2005, 71, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.M.; Bibby, K.J.; Lipus, D.; Hammack, R.W.; Gregory, K.B. The functional potential of microbial communities in hydraulic fracturing source water and produced water from natural gas extraction characterized by metagenomic sequencing. PLoS ONE 2014, 9, e107682. [Google Scholar] [CrossRef]
- Zhang, F.; She, Y.-H.; Chai, L.-J.; Banat, I.M.; Zhang, X.-T.; Shu, F.-C.; Wang, Z.-L.; Yu, L.-J.; Hou, D.-J. Microbial diversity in long-term water-flooded oil reservoirs with different in situ temperatures in China. Sci. Rep. 2012, 2, 760. [Google Scholar] [CrossRef] [PubMed]
- Ragkousi, K.; Eichenberger, P.; Van Ooij, C.; Setlow, P. Identification of a new gene essential for germination of Bacillus subtilis spores with Ca2+-dipicolinate. J. Bacteriol. 2003, 185, 2315–2329. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Wu, G. Can Biofilm be reversed through quorum sensing in Pseudomonas aeruginosa? Front. Microbiol. 2019, 10, 1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mand, J.; Enning, D. Oil field microorganisms cause highly localized corrosion on chemically inhibited carbon steel. Microb. Biotechnol. 2021, 14, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ma, S.; Hu, H.; Ding, L.; Ren, H. The biological role of N-acyl-homoserine lactone-based quorum sensing (QS) in EPS production and microbial community assembly during anaerobic granulation process. Sci. Rep. 2018, 8, 15793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsek, M.R.; Greenberg, E.P. Acyl-homoserine lactone quorum sensing in gram-negative bacteria: A signaling mechanism involved in associations with higher organisms. Proc. Natl. Acad. Sci. USA 2000, 97, 8789–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, R.; Vanderleyden, J.; Michiels, J. Quorum sensing and swarming migration in bacteria. FEMS Microbiol. Rev. 2004, 28, 261–289. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cheng, J.; Wang, Y.; Shen, X. The Pseudomonas quinolone signal (PQS): Not just for quorum sensing anymore. Front. Cell. Infect. Microbiol. 2018, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Almblad, H.; Harrison, J.J.; Rybtke, M.; Groizeleau, J.; Givskov, M.; Parsek, M.R.; Tolker-Nielsen, T. The cyclic AMP-Vfr signaling pathway in Pseudomonas aeruginosa is inhibited by cyclic Di-GMP. J. Bacteriol. 2015, 197, 2190–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, L.; Forest, K.T.; Maier, B. Type IV pili: Dynamics, biophysics and functional consequences. Nat. Rev. Microbiol. 2019, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Belas, R. Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol. 2014, 22, 517–527. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bin Name | MAG 1 | MAG 2 | MAG 3 | MAG 4 | MAG 5 | MAG 6 | MAG 7 | MAG 8 | MAG 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sequence statistics | Total length (bp) | 1,452,146 | 3,984,249 | 1,369,581 | 2,973,006 | 2,998,729 | 2,088,230 | 2,003,357 | 3,539,121 | 1,490,822 |
| Number of contigs | 312 | 327 | 217 | 100 | 96 | 227 | 185 | 151 | 293 | |
| N50 | 4863 | 19,895 | 7432 | 52,585 | 79,451 | 12,403 | 15,484 | 33,161 | 5366 | |
| GC content (%) | 29.33 | 69.87 | 42.84 | 58.14 | 41.21 | 33.17 | 33.80 | 43.35 | 30.18 | |
| Completion (%) | 76.06 | 98.59 | 76.32 | 92.96 | 97.18 | 59.15 | 100.00 | 98.59 | 47.89 | |
| Redundancy (%) | 2.82 | 7.04 | 6.58 | 2.82 | 2.82 | 5.63 | 1.41 | 1.41 | 2.82 | |
| Taxonomic assignation | Domain | Bacteria | Bacteria | Archaea | Bacteria | Bacteria | Bacteria | Bacteria | Bacteria | Bacteria |
| Phylum | Thermotogae | Proteobacteria | Euryarchaeota | Proteobacteria | Deferribacteres | Firmicutes | Firmicutes | Bacteroidetes | Thermotogae | |
| Class | Thermotogae | Gammaproteobacteria | Methanomicrobia | Gammaproteobacteria | Deferribacteres | Clostridia | Clostridia | Bacteroidia | Thermotogae | |
| Order | Petrotogales | Chromatiales | Methanosarcinales | Alteromonadales | Deferribacterales | Halanaerobiales | Halanaerobiales | Bacteroidales | Petrotogales | |
| Family | Petrotogaceae | Ectothiodospiraceae | Methanosarcinaceae | Alteromonadaceae | Defferibacteraceae | Halanaerobiaceae | Halanaerobiaceae | BM520 | Petrotogaceae | |
| Genus | Geotoga | Arhodomonas | Methanohalophilus | Marinobacter | Flexistipes | Halanaerobium | QPJE01 | - | Geotoga | |
| Species | G. petraea | A. aquaeolei | M. euhalobius | M. persicus | F. sinusarabici | H. congolense | sp003337245 | - | G. petreae |
| Bin Name | MAG 1 | MAG 2 | MAG 3 | MAG 4 | MAG 5 | MAG 6 | MAG 7 | MAG 8 | MAG 9 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Taxonomy | Geotoga petraea | Arhodomonas aquaeolei | Methanohalophilus euhalobius | Marinobacter persicus | Flexistipes sinusarabici | Halanaerobium congolense | QPJE01 | BM520 | Geotoga petreae | ||
| Completion (%) | 76.06 | 98.59 | 76.32 | 92.96 | 97.18 | 59.15 | 100.00 | 98.59 | 47.89 | ||
| Metabolism | Reaction | ||||||||||
| Fermentation | Acetate production (Wood-Ljungdahl pathway) | ||||||||||
| Succinate production (glyoxylate cycle) | |||||||||||
| Hydrogen production | |||||||||||
| Ethanol production | |||||||||||
| Peptide degradation | |||||||||||
| Hydrocarbondegradation | Benzene/toluene degradation | ||||||||||
| Methanogenesis | Methylotrophy | ||||||||||
| Sulfur metabolism | thiosulfate oxidation | ||||||||||
| thiosulfate reduction | |||||||||||
| Nitrogen metabolism | Dissimilatory nitrate reduction | ||||||||||
| Denitrification | |||||||||||
| Response to environment | Salt-in | ||||||||||
| Salt-out | |||||||||||
| Radiation resistance | |||||||||||
| Mg/Ca acquisition | |||||||||||
| Arsenate reduction | |||||||||||
| Endosporulation | |||||||||||
| Quorum sensing | |||||||||||
| Biofilm | |||||||||||
| Metabolic potential: | |||||||||||
| Present | |||||||||||
| Absent | |||||||||||
| Pathway completeness: | |||||||||||
| 100% | |||||||||||
| 50–99% | |||||||||||
| 1–49% | |||||||||||
| 0% | |||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheffer, G.; Hubert, C.R.J.; Enning, D.R.; Lahme, S.; Mand, J.; de Rezende, J.R. Metagenomic Investigation of a Low Diversity, High Salinity Offshore Oil Reservoir. Microorganisms 2021, 9, 2266. https://doi.org/10.3390/microorganisms9112266
Scheffer G, Hubert CRJ, Enning DR, Lahme S, Mand J, de Rezende JR. Metagenomic Investigation of a Low Diversity, High Salinity Offshore Oil Reservoir. Microorganisms. 2021; 9(11):2266. https://doi.org/10.3390/microorganisms9112266
Chicago/Turabian StyleScheffer, Gabrielle, Casey R. J. Hubert, Dennis R. Enning, Sven Lahme, Jaspreet Mand, and Júlia R. de Rezende. 2021. "Metagenomic Investigation of a Low Diversity, High Salinity Offshore Oil Reservoir" Microorganisms 9, no. 11: 2266. https://doi.org/10.3390/microorganisms9112266
APA StyleScheffer, G., Hubert, C. R. J., Enning, D. R., Lahme, S., Mand, J., & de Rezende, J. R. (2021). Metagenomic Investigation of a Low Diversity, High Salinity Offshore Oil Reservoir. Microorganisms, 9(11), 2266. https://doi.org/10.3390/microorganisms9112266