
Factors Driving Bacterial Microbiota of Eggs from Commercial Hatcheries of European Seabass and Gilthead Seabream


 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Broodstock Culture Conditions and Disinfection Protocol
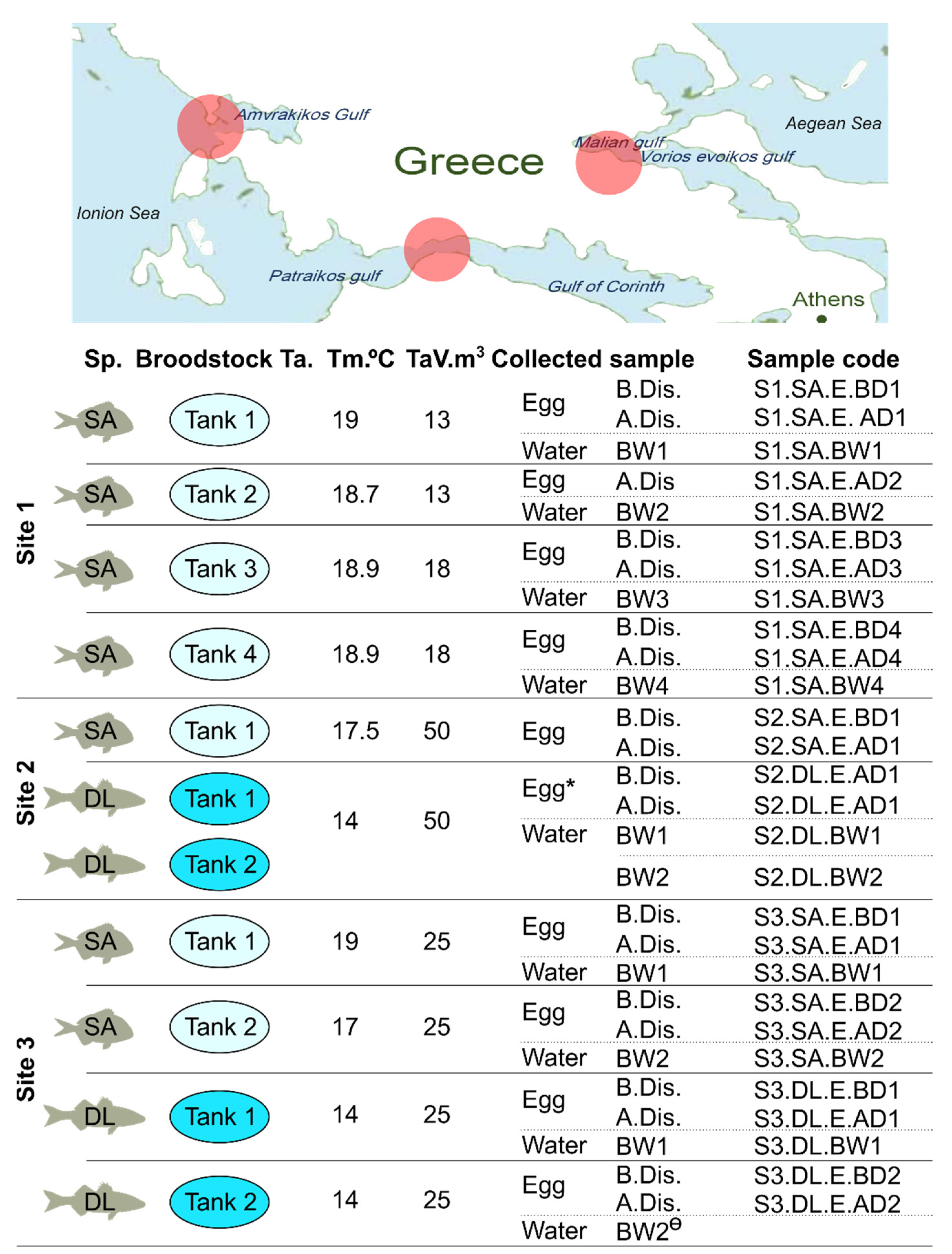
2.2. Sample Collection
2.3. DNA Extraction
2.4. 16S rRNA Library Construction and Sequencing
2.5. Sequence Processing and Bioinformatics
2.6. Functional Analysis
2.7. Quantitative Analysis of 16S rRNA Gene
2.8. Statistics
3. Results
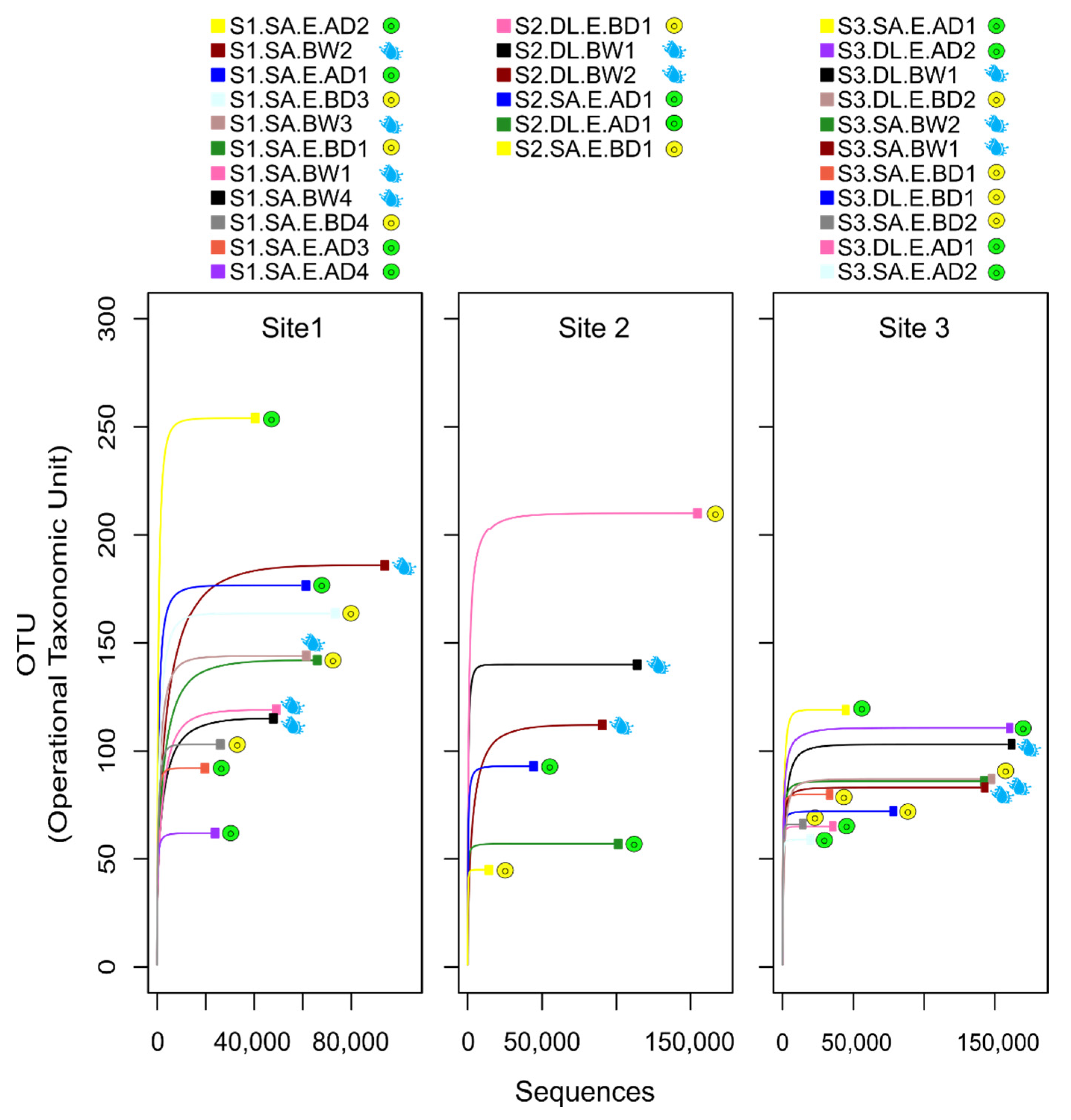
3.1. Sequencing and Rarefaction Outcome
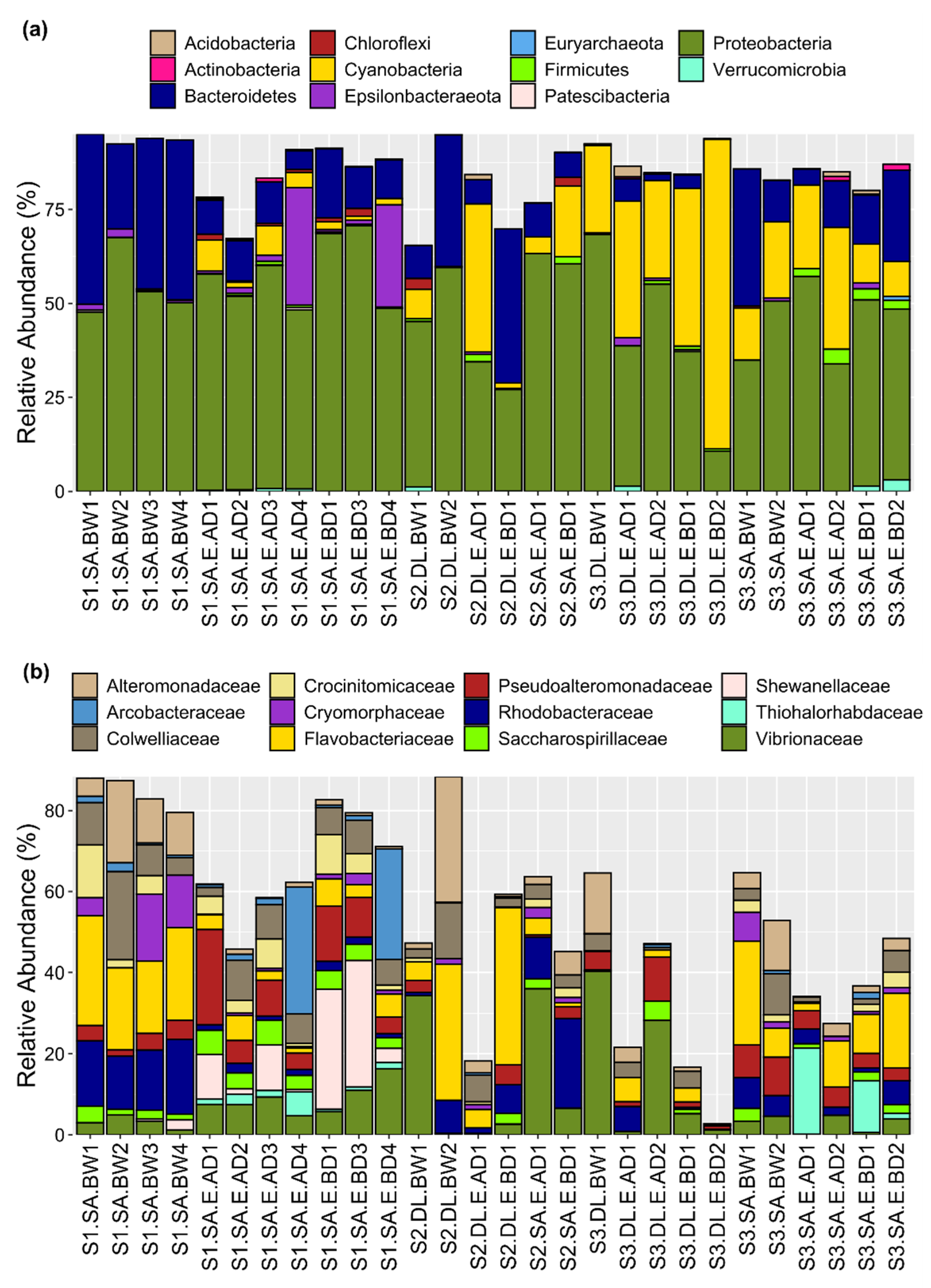
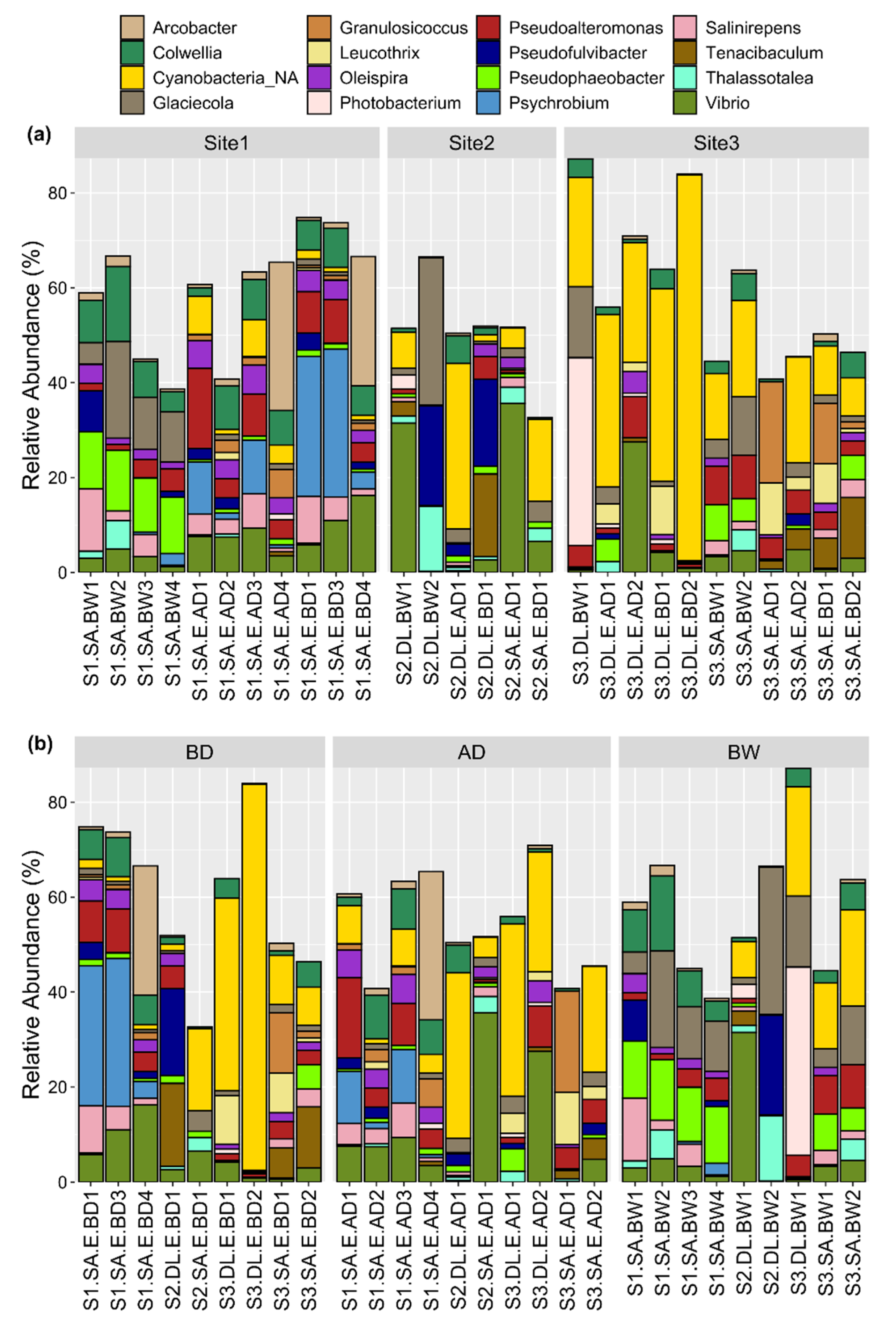
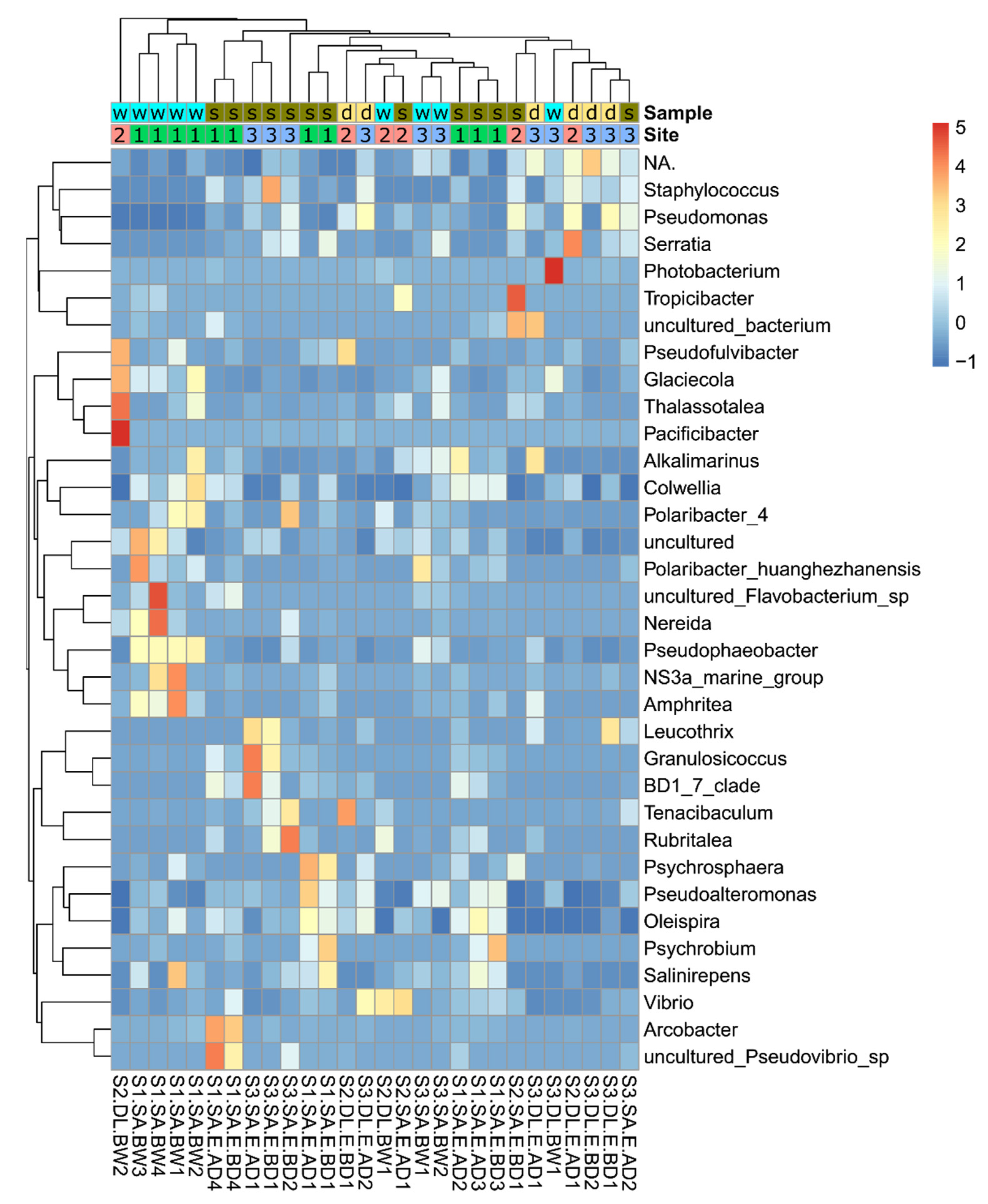
3.2. Bacterial Community Taxonomic Composition
3.3. Bacterial Community Diversity
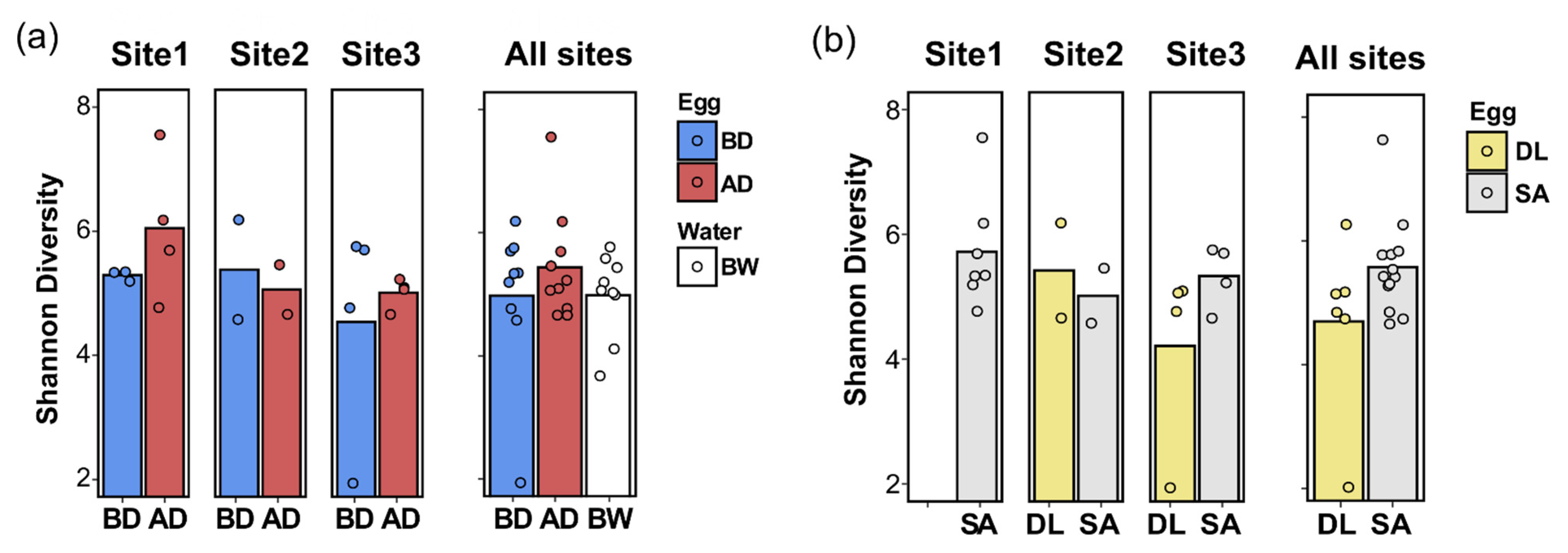
3.3.1. Alpha-Diversity
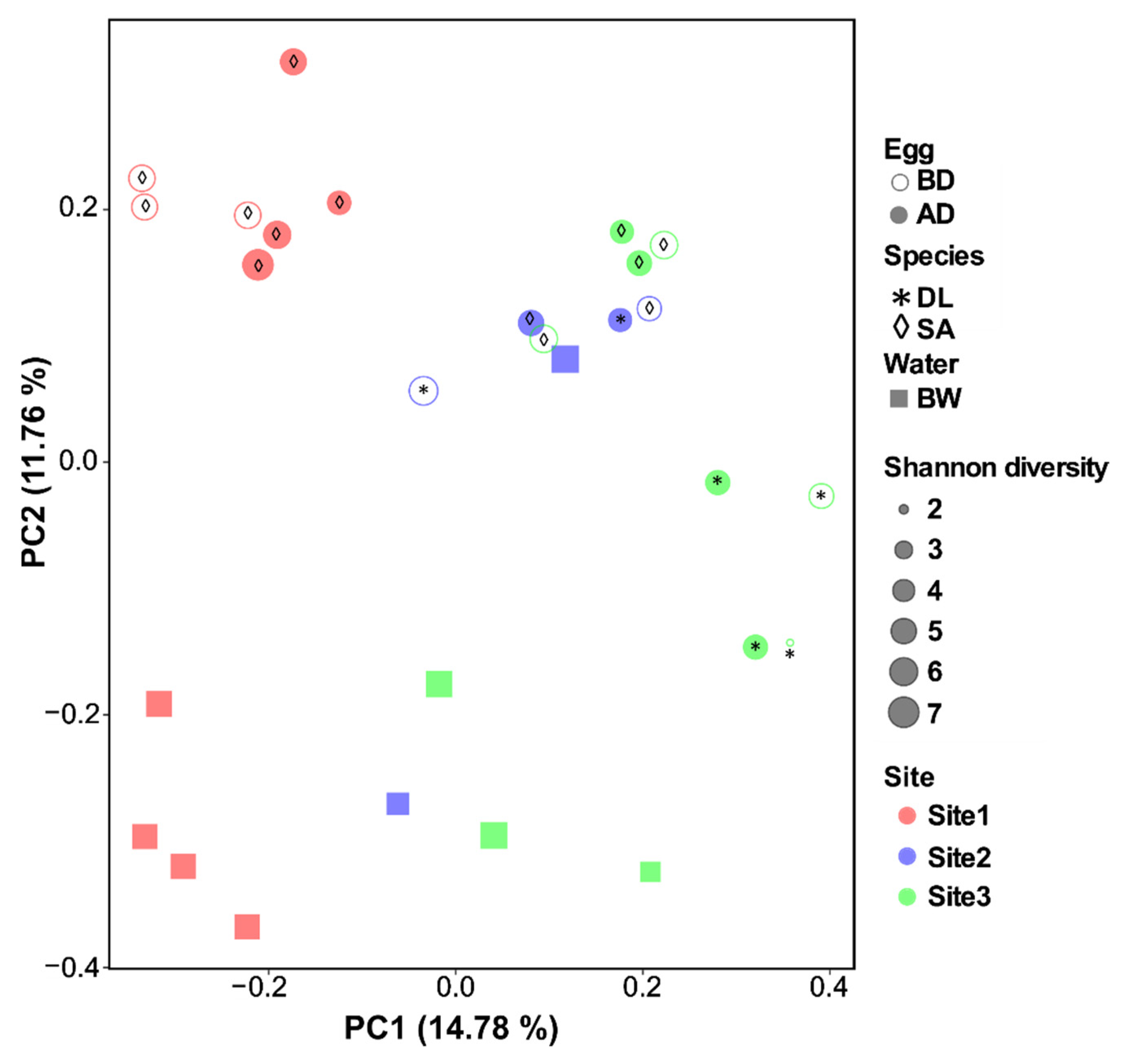
3.3.2. Beta-Diversity
3.4. Relative Abundance
3.5. Functional Prediction
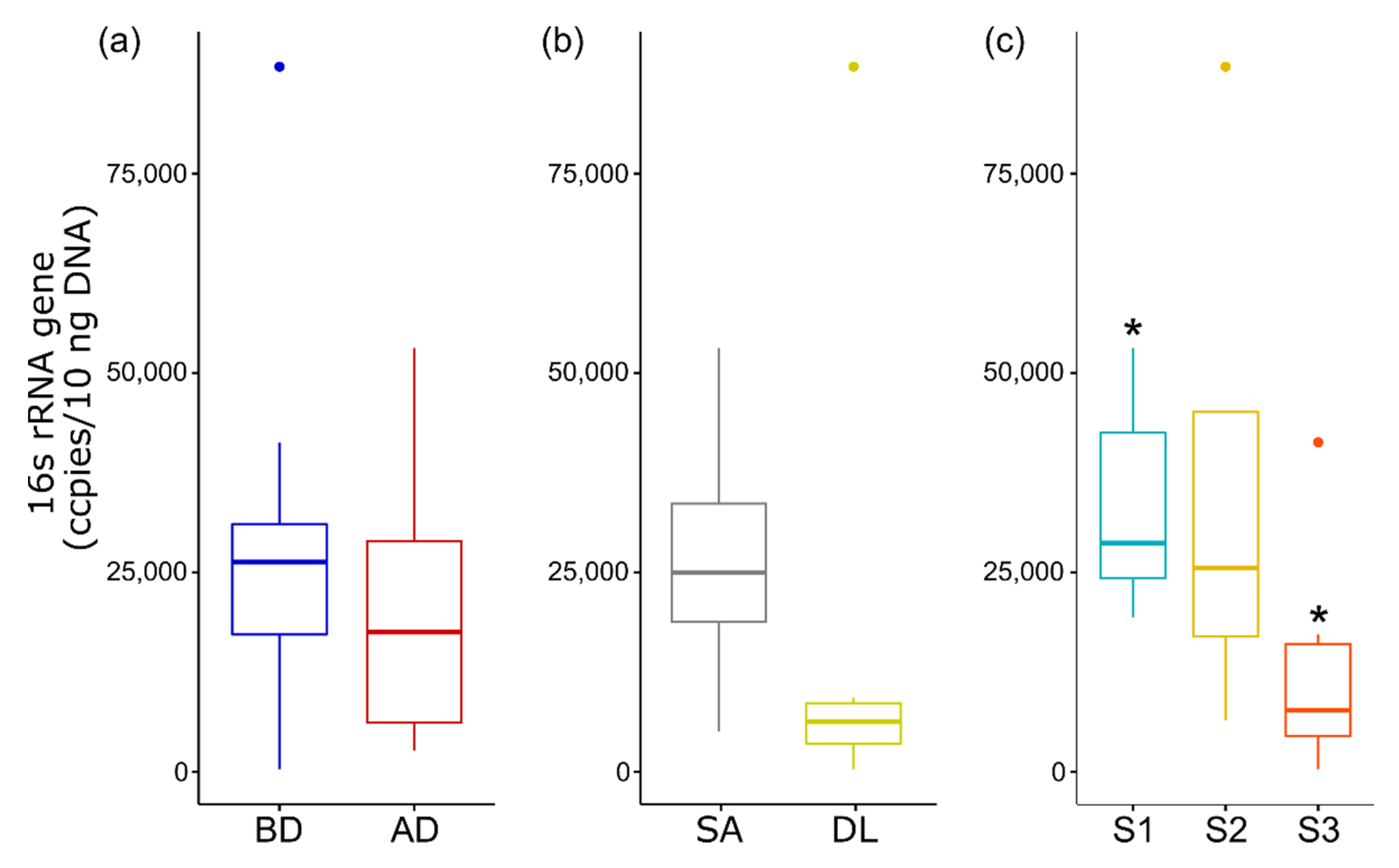
3.6. Quantitative Analysis of 16S rRNA Gene
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilbert, S.F.; Sapp, J.; Tauber, A.I. A symbiotic view of life: We have never been individuals. Q. Rev. Biol. 2012, 87, 325–341. [Google Scholar] [CrossRef]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Lošo, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef]
- Proctor, L.M.; Creasy, H.H.; Fettweis, J.M.; Lloyd-Price, J.; Mahurkar, A.; Zhou, W.; Buck, G.A.; Snyder, M.P.; Strauss, J.F.; Weinstock, G.M.; et al. The Integrative human microbiome project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef]
- Hadrich, D. Microbiome research is becoming the key to better understanding health and nutrition. Front. Genet. 2018, 9, 212. [Google Scholar] [CrossRef]
- Brugman, S.; Ikeda-Ohtsubo, W.; Braber, S.; Folkerts, G.; Pieterse, C.M.J.; Bakker, P.A.H.M. A Comparative review on microbiota manipulation: Lessons from fish, plants, livestock, and human research. Front. Nutr. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Simon, C.; Daniel, R. Metagenomic analyses: Past and future trends. Appl. Environ. Microbiol. 2011, 77, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Groff, J.M. Cutaneous biology and diseases of fish. Vet. Clin. North Am. Exot. Anim. Pract. 2001, 4, 321–411. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5, 1. [Google Scholar] [CrossRef]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The gut microbiota of marine fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef]
- Legrand, T.P.R.A.; Catalano, S.R.; Wos-Oxley, M.L.; Stephens, F.; Landos, M.; Bansemer, M.S.; Stone, D.A.J.; Qin, J.G.; Oxley, A.P.A. The inner workings of the outer surface: Skin and gill microbiota as indicators of changing gut health in yellowtail kingfish. Front. Microbiol. 2018, 8, 2664. [Google Scholar] [CrossRef]
- Legrand, T.P.R.A.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P.A. A microbial sea of possibilities: Current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 2020, 12, 1101–1134. [Google Scholar] [CrossRef]
- Tellez, G.; Higgins, S.E.; Donoghue, A.M.; Hargis, B.M. Digestive physiology and the role of microorganisms. J. Appl. Poult. Res. 2006, 15, 136–144. [Google Scholar] [CrossRef]
- Newaj-Fyzul, A.; Al-Harbi, A.H.; Austin, B. Review: Developments in the use of probiotics for disease control in aquaculture. Aquaculture 2014, 431, 1–11. [Google Scholar] [CrossRef]
- Ringø, E.; Dimitroglou, A.; Hoseinifar, S.H.; Davies, S.J. Prebiotics in finfish: An update. In Aquaculture Nutrition: Gut Health, Probiotics and Prebiotics; Merrifield, D.L., RingØ, E., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2014; pp. 360–400. [Google Scholar]
- Wang, A.R.; Ran, C.; Ringø, E.; Zhou, Z.G. Progress in fish gastrointestinal microbiota research. Rev. Aquac. 2018, 10, 626–640. [Google Scholar] [CrossRef]
- Hansen, G.H.; Olafsen, J.A. Bacterial interactions in early life stages of marine cold water fish. Microb. Ecol. 1999, 38, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Olafsen, J.A. Interactions between fish larvae and bacteria in marine aquaculture. Aquaculture 2001, 200, 223–247. [Google Scholar] [CrossRef]
- Merrifield, D.L.; Rodiles, A. The fish microbiome and its interactions with mucosal tissues. In Mucosal Health Aquaculture; Beck, B.H., Peatman, E., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 273–295. [Google Scholar]
- Borges, N.; Keller-Costa, T.; Sanches-Fernandes, G.M.M.; Louvado, A.; Gomes, N.C.M.; Costa, R. Bacteriome structure, function, and probiotics in fish larviculture: The good, the bad, and the gaps. Annu. Rev. Anim. Biosci. 2021, 9, 423–452. [Google Scholar] [CrossRef]
- Vadstein, O.; Bergh, Ø.; Gatesoupe, F.-J.; Galindo-Villegas, J.; Mulero, V.; Picchietti, S.; Scapigliati, G.; Makridis, P.; Olsen, Y.; Dierckens, K.; et al. Microbiology and immunology of fish larvae. Rev. Aquac. 2013, 5, S1–S25. [Google Scholar] [CrossRef]
- Abdul Razak, S.; Griffin, M.J.; Mischke, C.C.; Bosworth, B.G.; Waldbieser, G.C.; Wise, D.J.; Marsh, T.L.; Scribner, K.T. Biotic and abiotic factors influencing channel catfish egg and gut microbiome dynamics during early life stages. Aquaculture 2019, 498, 556–567. [Google Scholar] [CrossRef]
- Bone, A.; Bekaert, M.; Papadopoulou, A.; McMillan, S.; Adams, A.; Davie, A.; Desbois, A.P. Bacterial communities of ballan wrasse (Labrus bergylta) eggs at a commercial marine hatchery. Curr. Microbiol. 2020, 1, 3. [Google Scholar] [CrossRef]
- FEAP. Federation of European Aquaculture Producers Annual Report. Available online: https://feap.info/index.php/data/ (accessed on 27 December 2020).
- Muniesa, A.; Basurco, B.; Aguilera, C.; Furones, D.; Reverté, C.; Sanjuan-Vilaplana, A.; Jansen, M.D.; Brun, E.; Tavornpanich, S. Mapping the knowledge of the main diseases affecting sea bass and sea bream in Mediterranean. Transbound. Emerg. Dis. 2020, 67, 1089–1100. [Google Scholar] [CrossRef]
- Yoder, M.; De Ley, I.T.; King, I.W.; Mundo-Ocampo, M.; Mann, J.; Blaxter, M.; Poiras, L.; Ley, P. De DESS: A versatile solution for preserving morphology and extractable DNA of nematodes. Nematology 2006, 8, 367–376. [Google Scholar] [CrossRef]
- Pinto, P.I.S.; Guerreiro, C.C.; Costa, R.A.; Martinez-Blanch, J.F.; Carballo, C.; Codoñer, F.M.; Manchado, M.; Power, D.M. Understanding pseudo-albinism in sole (Solea senegalensis): A transcriptomics and metagenomics approach. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 January 2020).
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using Microbiome Analyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; Van de Geer, S.; De Kort, F.; van Houwellingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Califano, G.; Castanho, S.; Soares, F.; Ribeiro, L.; Cox, C.J.; Mata, L.; Costa, R. Molecular taxonomic profiling of bacterial communities in a Gilthead Seabream (Sparus aurata) hatchery. Front. Microbiol. 2017, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Tan, K.; Gong, P.; Lei, P.; Guo, Z.; Wang, S.; Gao, S.; Zhou, Y.; Shu, Y.; Zhou, X.; et al. Correlation of microbiota in the gut of fish species and water. 3 Biotech 2020, 10, 472. [Google Scholar] [CrossRef]
- Silveira, S.B.; Odebrecht, C. Effects of salinity and temperature on the growth, toxin production, and akinete germination of the Cyanobacterium Nodularia Spumigena. Front. Mar. Sci. 2019, 6, 339. [Google Scholar] [CrossRef]
- Lokesh, J.; Kiron, V. Transition from freshwater to seawater reshapes the skin-associated microbiota of Atlantic salmon. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Wilkins, L.G.E.; Rogivue, A.; Schütz, F.; Fumagalli, L.; Wedekind, C. Increased diversity of egg-associated bacteria on brown trout (Salmo trutta) at elevated temperatures. Sci. Rep. 2015, 5, 17084. [Google Scholar] [CrossRef] [PubMed]
- Roalkvam, I.; Drønen, K.; Dahle, H.; Wergeland, H.I. Microbial communities in a flow-through fish farm for lumpfish (Cyclopterus lumpus L.) during healthy rearing conditions. Front. Microbiol. 2019, 10, 1594. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, L.G.E.; Fumagalli, L.; Wedekind, C. Effects of host genetics and environment on egg-associated microbiotas in brown trout (Salmo trutta). Mol. Ecol. 2016, 25, 4930–4945. [Google Scholar] [CrossRef]
- Paix, B.; Carriot, N.; Barry-Martinet, R.; Greff, S.; Misson, B.; Briand, J.F.; Culioli, G. A multi-omics analysis suggests links between the differentiated surface metabolome and epiphytic microbiota along the thallus of a Mediterranean Seaweed holobiont. Front. Microbiol. 2020, 11, 494. [Google Scholar] [CrossRef]
- Uren Webster, T.M.; Consuegra, S.; Hitchings, M.; Garcia de Leaniz, C. Interpopulation variation in the Atlantic salmon microbiome reflects environmental and genetic diversity. Appl. Environ. Microbiol. 2018, 84, e00691-18. [Google Scholar] [CrossRef] [PubMed]
- Migaud, H.; Bell, G.; Cabrita, E.; Mcandrew, B.; Davie, A.; Bobe, J.; Herráez, M.P.; Carrillo, M. Gamete quality and broodstock management in temperate fish. Rev. Aquac. 2013, 5, S194–S223. [Google Scholar] [CrossRef]
- Van Vliet, D.; Loch, T.P.; Faisal, M. Flavobacterium psychrophilum infections in salmonid broodstock and hatchery-propagated stocks of the Great Lakes basin. J. Aquat. Anim. Health 2015, 27, 192–202. [Google Scholar] [CrossRef]
- Pradeep, P.J.; Suebsing, R.; Sirthammajak, S.; Kampeera, J.; Jitrakorn, S.; Saksmerprome, V.; Turner, W.; Palang, I.; Vanichviriyakit, R.; Senapin, S.; et al. Evidence of vertical transmission and tissue tropism of Streptococcosis from naturally infected red tilapia (Oreochromis spp.). Aquac. Reports 2016, 3, 58–66. [Google Scholar] [CrossRef]
- De Swaef, E.; Van den Broeck, W.; Dierckens, K.; Decostere, A. Disinfection of teleost eggs: A review. Rev. Aquac. 2016, 8, 321–341. [Google Scholar] [CrossRef]
- Can, E.; Saka, Ş.; Firat, M.K. Disinfection of gilthead sea bream (Sparus aurata), red porgy (Pagrus pagrus), and common Dentex (Dentex dentex) eggs from Sparidae with different disinfectants. Kafkas Üniversitesi Vet. Fakültesi Derg. 2010, 16, 299–306. [Google Scholar]
- McDonnell, G.; Russell, A.D. Antiseptics and disinfectants: Activity, action, and resistance. Clin. Microbiol. Rev. 1999, 12, 147. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.; Eissa, A.E.; Hanna, M.; Okada, M.A. Identifying some pathogenic Vibrio/Photobacterium species during mass mortalities of cultured Gilthead seabream (Sparus aurata) and European seabass (Dicentrarchus labrax) from some Egyptian coastal provinces. Int. J. Vet. Sci. Med. 2013, 1, 87–95. [Google Scholar] [CrossRef]
- Johnson, P.W.; Sieburth, J.M.; Sastry, A.; Arnold, C.R.; Doty, M.S. Leucothrix mucor infestation of benthic crustacea, fish eggs, and tropical algae. Limnol. Oceanogr. 1971, 16, 962–969. [Google Scholar] [CrossRef]
- Hansen, G.H.; Olafsen, J.A. Bacterial colonization of cod (Gadus morhua L.) and halibut (Hippoglossus hippoglossus) eggs in marine aquaculture. Appl. Environ. Microbiol. 1989, 55, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Fusco, V.; Quero, G.M. Nucleic acid-based methods to identify, detect and type pathogenic bacteria occurring in milk and dairy products. In Structure and Function of Food Engineering; Ayman, A.E., Ed.; Tech–Open Access Company: Rijeka, Croatia, 2012; pp. 371–404. [Google Scholar] [CrossRef]
- Lazcka, O.; Del Campo, F.J.; Munoz, F.X. Pathogen detection: A perspective of traditional methods and biosensors. Biosens. Bioelectron. 2007, 22, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Metwally, N.H.; Mohamed, M.S. New imidazolone derivatives comprising a benzoate or sulfonamide moiety as anti-inflammatory and antibacterial inhibitors: Design, synthesis, selective COX-2, DHFR and molecular-modeling study. Bioorg. Chem. 2020, 99, 103438. [Google Scholar] [CrossRef]
- Kim, J.M.; Marshall, M.R.; Cornell, J.A.; Iii, J.P.; Wei, C.I. Antibacterial activity of carvacrol, citral, and geraniol against Salmonella typhimurium in culture medium and on fish cubes. J. Food Sci. 1995, 60, 1364–1368. [Google Scholar] [CrossRef]
- Pontes, E.K.U.; Melo, H.M.; Nogueira, J.W.A.; Firmino, N.C.S.; de Carvalho, M.G.; Catunda Júnior, F.E.A.; Cavalcante, T.T.A. Antibiofilm activity of the essential oil of citronella (Cymbopogon nardus) and its major component, geraniol, on the bacterial biofilms of Staphylococcus aureus. Food Sci. Biotechnol. 2019, 28, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Höschle, B.; Jendrossek, D. Utilization of geraniol is dependent on molybdenum in Pseudomonas aeruginosa: Evidence for different metabolic routes for oxidation of geraniol and citronellol. Microbiology 2005, 151, 2277–2283. [Google Scholar] [CrossRef]
- Uhlik, O.; Wald, J.; Strejcek, M.; Musilova, L.; Ridl, J.; Hroudova, M.; Vlcek, C.; Cardenas, E.; Mackova, M.; Macek, T. Identification of bacteria utilizing biphenyl, benzoate, and naphthalene in long-term contaminated soil. PLoS ONE 2012, 7, e40653. [Google Scholar] [CrossRef] [PubMed]
- Baxi, N.N. Influence of ε-caprolactam on growth and physiology of environmental bacteria. Ann. Microbiol. 2013, 63, 1471–1476. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | BS | Species | Tank | Male (n) | Female (n) | Density (kg/m3) | Weight ֎ (kg) |
|---|---|---|---|---|---|---|---|
| 1 | BS 1 | SA | 1 | 10 | 31 | 8.36 | 2.02 |
| 1 | BS 2 | SA | 2 | 11 | 20 | 5.28 | 2.21 |
| 1 | BS 3 | SA | 3 | 16 | 30 | 5.82 | 2.23 |
| 1 | BS 4 | SA | 4 | 10 | 27 | 6.78 | 3.01 |
| 2 | BS 1 | SA | 1 | 38 | 105 | 5.9 | 1.90 |
| 2 | BS 1 | DL | 1 | 67 | 68 | 14.9 | 5.00 |
| 2 | BS 2 | DL | 2 | 50 | 75 | 7.7 | 2.80 |
| 3 | BS 1 | SA | 1 | 63 | 20 | 5.29 | 1.59 |
| 3 | BS 2 | SA | 2 | 23 | 25 | 3.45 | 1.79 |
| 3 | BS 1 | DL | 1 | 23 | 28 | 7.11 | 3.48 |
| 3 | BS 2 | DL | 2 | 25 | 31 | 8.61 | 3.81 |
| Df | Sum Sq | Mean Sq | F. Model | R2 | Pr (>F) | |
|---|---|---|---|---|---|---|
| Site | 1 | 1.2367 | 1.23667 | 3.9341 | 0.12130 | 0.000999 *** |
| Sample type (eggs vs. water) | 1 | 1.0998 | 1.09975 | 3.4986 | 0.10787 | 0.000999 *** |
| Residuals | 25 | 7.8586 | 0.31434 | 0.77083 | ||
| Total | 27 | 10.1950 | 1.00000 |
| Df | Sum Sq | Mean Sq | F. Model | R2 | Pr (>F) | |
|---|---|---|---|---|---|---|
| Site | 1 | 1.0757 | 1.07570 | 3.6486 | 0.16206 | 0.000999 *** |
| Species | 1 | 0.7011 | 0.70111 | 2.3780 | 0.10563 | 0.000999 *** |
| Disinfection | 1 | 0.1703 | 0.17031 | 0.5777 | 0.02566 | 0.962038 |
| Site: Species | 1 | 0.5628 | 0.56280 | 1.9089 | 0.08479 | 0.005994 ** |
| Residuals | 14 | 4.1276 | 0.29483 | 0.62186 | ||
| Total | 18 | 6.6375 | 1.00000 |
| Pathway | Site | Species | Type | Disinfection | All | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| KO | Name | S1 | S2 | S3 | Sa | Dl | E | W | B | A | |
| ko01230 | Biosynthesis of amino acids | 6.7 | 7.4 | 7.2 | 6.9 | 7.4 | 7.0 | 7.1 | 7.0 | 7.1 | 7.0 |
| ko01200 | Carbon metabolism | 6.9 | 6.7 | 6.6 | 6.7 | 6.6 | 6.7 | 6.8 | 6.7 | 6.7 | 6.7 |
| ko00230 | Purine metabolism | 4.3 | 4.7 | 4.5 | 4.3 | 4.5 | 4.4 | 4.6 | 4.5 | 4.3 | 4.5 |
| ko00240 | Pyrimidine metabolism | 3.1 | 3.3 | 3.3 | 3.1 | 3.4 | 3.2 | 3.4 | 3.3 | 3.1 | 3.2 |
| ko00620 | Pyruvate metabolism | 2.9 | 2.9 | 2.9 | 2.9 | 2.9 | 2.9 | 3.0 | 2.9 | 2.9 | 2.9 |
| ko00330 | Arginine and proline metabolism | 2.4 | 2.5 | 2.5 | 2.4 | 2.5 | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 |
| ko00260 | Glycine, serine, and threonine metabolism | 2.3 | 2.3 | 2.4 | 2.3 | 2.3 | 2.3 | 2.5 | 2.3 | 2.2 | 2.3 |
| ko00250 | Alanine, aspartate, and glutamate metabolism | 2.3 | 2.3 | 2.3 | 2.3 | 2.2 | 2.3 | 2.3 | 2.3 | 2.3 | 2.3 |
| ko00010 | Glycolysis/Gluconeogenesis | 2.1 | 2.2 | 2.4 | 2.1 | 2.4 | 2.2 | 2.3 | 2.2 | 2.2 | 2.2 |
| ko00720 | Carbon fixation pathways in prokaryotes | 2.2 | 2.3 | 2.2 | 2.2 | 2.3 | 2.2 | 2.3 | 2.2 | 2.2 | 2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najafpour, B.; Pinto, P.I.S.; Moutou, K.A.; Canario, A.V.M.; Power, D.M. Factors Driving Bacterial Microbiota of Eggs from Commercial Hatcheries of European Seabass and Gilthead Seabream. Microorganisms 2021, 9, 2275. https://doi.org/10.3390/microorganisms9112275
Najafpour B, Pinto PIS, Moutou KA, Canario AVM, Power DM. Factors Driving Bacterial Microbiota of Eggs from Commercial Hatcheries of European Seabass and Gilthead Seabream. Microorganisms. 2021; 9(11):2275. https://doi.org/10.3390/microorganisms9112275
Chicago/Turabian StyleNajafpour, Babak, Patricia I. S. Pinto, Katerina A. Moutou, Adelino V. M. Canario, and Deborah M. Power. 2021. "Factors Driving Bacterial Microbiota of Eggs from Commercial Hatcheries of European Seabass and Gilthead Seabream" Microorganisms 9, no. 11: 2275. https://doi.org/10.3390/microorganisms9112275
APA StyleNajafpour, B., Pinto, P. I. S., Moutou, K. A., Canario, A. V. M., & Power, D. M. (2021). Factors Driving Bacterial Microbiota of Eggs from Commercial Hatcheries of European Seabass and Gilthead Seabream. Microorganisms, 9(11), 2275. https://doi.org/10.3390/microorganisms9112275