
Overcoming the Prokaryote/Eukaryote Barrier in Tuberculosis Treatment: A Prospect for the Repurposing and Use of Antiparasitic Drugs


Abstract
1. Introduction
2. Classical Antiparasitic Drugs for Potential Anti-TB Treatment
2.1. Avermectins
2.2. Mefloquine
2.3. Niclosamide
2.4. Nitazoxanide
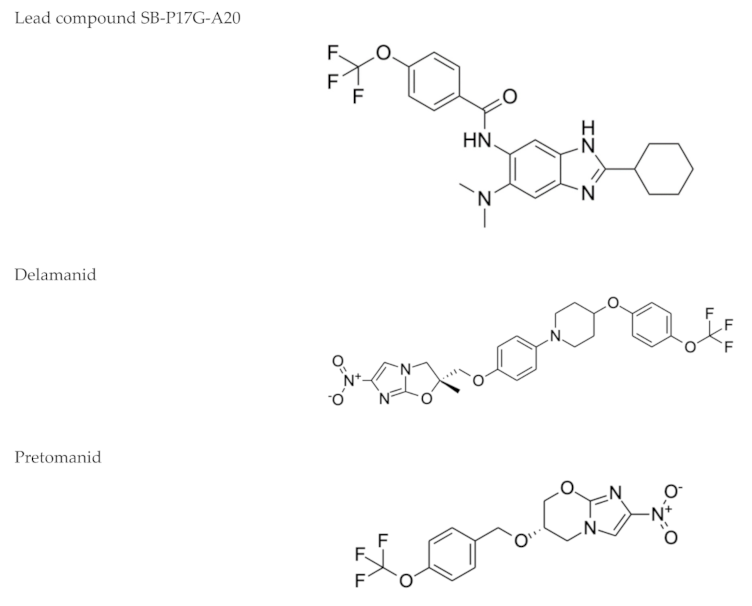
2.5. Nitroimidazoles
2.6. Pyronaridine
2.7. Auranofin
3. Plants as a Source of Natural Products Used in Traditional Medicine
4. A Special Case: The Dual Antiparasitic and Anti-TB Activity of Bacteriocin AS-48
5. Discussion
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- ECDC. Tackling the Burden in the European Union. Available online: https://www.oecd.org/health/health-systems/AMR-Tackling-the-Burden-in-the-EU-OECD-ECDC-Briefing-Note-2019.pdf (accessed on 13 October 2021).
- ECDC. Antimicrobial Resistance. Available online: https://www.ecdc.europa.eu/en/antimicrobial-resistance (accessed on 13 October 2021).
- ECDC. Antimicrobial Resistance in the EU/EEA (EARS-Net)—Annual Epidemiological Report for 2019. Available online: https://www.ecdc.europa.eu/en/publications-data/surveillance-antimicrobial-resistance-europe-2019 (accessed on 13 October 2021).
- CDC. Antibiotic/Antimicrobial Resistance (AR/AMR). Available online: https://www.cdc.gov/drugresistance/index.html (accessed on 13 October 2021).
- O’Neill, J. Review on Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Available online: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (accessed on 13 October 2021).
- O’Neill, J. Review on Antimicrobial Resistance: Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 13 October 2021).
- Jackson, N.; Czaplewski, L.; Piddock, L.J.V. Discovery and development of new antibacterial drugs: Learning from experience? J. Antimicrob. Chemother. 2018, 73, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Gajdacs, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Conly, J.; Johnston, B. Where are all the new antibiotics? The new antibiotic paradox. Can. J. Infect. Dis. Med. Microbiol. 2005, 16, 159–160. [Google Scholar] [CrossRef] [PubMed]
- CBO. Research and Development in the Pharmaceutical Industry. Available online: https://www.cbo.gov/publication/57126 (accessed on 13 October 2021).
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic discovery: History, methods and perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef]
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Lienhardt, C.R.; Raviglione, M.C. TB Elimination Requires Discovery and Development of Transformational Agents. Appl. Sci. 2020, 10, 2605. [Google Scholar] [CrossRef]
- WHO. WHO Announces Updated Definitions of Extensively Drug-Resistant Tuberculosis. Available online: https://www.who.int/news/item/27-01-2021-who-announces-updated-definitions-of-extensively-drug-resistant-tuberculosis (accessed on 13 October 2021).
- Schein, C.H. Repurposing approved drugs on the pathway to novel therapies. Med. Res. Rev. 2020, 40, 586–605. [Google Scholar] [CrossRef]
- Farha, M.A.; Brown, E.D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 2019, 4, 565–577. [Google Scholar] [CrossRef]
- Kigondu, E.M.; Wasuna, A.; Warner, D.F.; Chibale, K. Pharmacologically active metabolites, combination screening and target identification-driven drug repositioning in antituberculosis drug discovery. Bioorg. Med. Chem. 2014, 22, 4453–4461. [Google Scholar] [CrossRef]
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24, 4373. [Google Scholar] [CrossRef]
- Zimmermann, P.; Curtis, N. Antimicrobial Effects of Antipyretics. Antimicrob. Agents Chemother. 2017, 61, e02268-16. [Google Scholar] [CrossRef]
- Firth, A.; Prathapan, P. Azithromycin: The First Broad-spectrum Therapeutic. Eur. J. Med. Chem. 2020, 207, 112739. [Google Scholar] [CrossRef]
- Fan, M.; Chen, S.; Weng, Y.; Li, X.; Jiang, Y.; Wang, X.; Bie, M.; An, L.; Zhang, M.; Chen, B.; et al. Ciprofloxacin promotes polarization of CD86+CD206 macrophages to suppress liver cancer. Oncol. Rep. 2020, 44, 91–102. [Google Scholar] [CrossRef]
- Burg, R.W.; Miller, B.M.; Baker, E.E.; Birnbaum, J.; Currie, S.A.; Hartman, R.; Kong, Y.L.; Monaghan, R.L.; Olson, G.; Putter, I.; et al. Avermectins, new family of potent anthelmintic agents: Producing organism and fermentation. Antimicrob. Agents Chemother. 1979, 15, 361–367. [Google Scholar] [CrossRef]
- Omura, S.; Crump, A. The life and times of ivermectin—A success story. Nat. Rev. Microbiol. 2004, 2, 984–989. [Google Scholar] [CrossRef]
- Chabala, J.C.; Mrozik, H.; Tolman, R.L.; Eskola, P.; Lusi, A.; Peterson, L.H.; Woods, M.F.; Fisher, M.H.; Campbell, W.C.; Egerton, J.R.; et al. Ivermectin, a new broad-spectrum antiparasitic agent. J. Med. Chem. 1980, 23, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Egerton, J.R.; Ostlind, D.A.; Blair, L.S.; Eary, C.H.; Suhayda, D.; Cifelli, S.; Riek, R.F.; Campbell, W.C. Avermectins, new family of potent anthelmintic agents: Efficacy of the B1a component. Antimicrob. Agents Chemother. 1979, 15, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Ward, S. The Nobel Prize in Medicine 2015: Two drugs that changed global health. Sci. Transl. Med. 2015, 7, 316ed14. [Google Scholar] [CrossRef]
- WHO. Lymphatic Filariasis. Available online: https://www.who.int/news-room/fact-sheets/detail/lymphatic-filariasis (accessed on 13 October 2021).
- WHO. Onchocerciasis. Available online: https://www.who.int/news-room/fact-sheets/detail/onchocerciasis (accessed on 13 October 2021).
- ACS. Discovery of Ivermectin. Available online: https://www.acs.org/content/dam/acsorg/education/whatischemistry/landmarks/discovery-of-ivermectin-mectizan.pdf (accessed on 13 October 2021).
- Laing, R.; Gillan, V.; Devaney, E. Ivermectin—Old Drug, New Tricks? Trends Parasitol. 2017, 33, 463–472. [Google Scholar] [CrossRef] [PubMed]
- The Ivermectin Roadmappers; Billingsley, P.; Binka, F.; Chaccour, C.; Foy, B.; Gold, S.; Gonzalez-Silva, M.; Jacobson, J.; Jagoe, G.; Jones, C.; et al. A Roadmap for the Development of Ivermectin as a Complementary Malaria Vector Control Tool. Am. J. Trop Med. Hyg. 2020, 102, 3–24. [Google Scholar] [CrossRef]
- Lim, L.E.; Vilcheze, C.; Ng, C.; Jacobs, W.R., Jr.; Ramon-Garcia, S.; Thompson, C.J. Anthelmintic avermectins kill Mycobacterium tuberculosis, including multidrug-resistant clinical strains. Antimicrob. Agents Chemother. 2013, 57, 1040–1046. [Google Scholar] [CrossRef]
- Muhammed Ameen, S.; Drancourt, M. Ivermectin lacks antituberculous activity. J. Antimicrob. Chemother. 2013, 68, 1936–1937. [Google Scholar] [CrossRef]
- Wolstenholme, A.J.; Rogers, A.T. Glutamate-gated chloride channels and the mode of action of the avermectin/milbemycin anthelmintics. Parasitology 2005, 131, S85–S95. [Google Scholar] [CrossRef]
- Youm, J.; Saier, M.H., Jr. Comparative analyses of transport proteins encoded within the genomes of Mycobacterium tuberculosis and Mycobacterium leprae. Biochim. Biophys. Acta 2012, 1818, 776–797. [Google Scholar] [CrossRef]
- Portaels, F.; Silva, M.T.; Meyers, W.M. Buruli ulcer. Clin. Derm. 2009, 27, 291–305. [Google Scholar] [CrossRef]
- Scherr, N.; Pluschke, G.; Thompson, C.J.; Ramon-Garcia, S. Selamectin Is the Avermectin with the Best Potential for Buruli Ulcer Treatment. PLoS Negl. Trop. Dis. 2015, 9, e0003996. [Google Scholar] [CrossRef] [PubMed]
- Omansen, T.F.; Porter, J.L.; Johnson, P.D.; van der Werf, T.S.; Stienstra, Y.; Stinear, T.P. In-vitro activity of avermectins against Mycobacterium ulcerans. PLoS Negl. Trop. Dis. 2015, 9, e0003549. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Munoz, L.; Shoen, C.; Sweet, G.; Vitoria, A.; Bull, T.J.; Cynamon, M.; Thompson, C.J.; Ramon-Garcia, S. Repurposing Avermectins and Milbemycins against Mycobacteroides abscessus and Other Nontuberculous Mycobacteria. Antibiotics 2021, 10, 381. [Google Scholar] [CrossRef] [PubMed]
- Palomino, J.C.; Martin, A. Is repositioning of drugs a viable alternative in the treatment of tuberculosis? J. Antimicrob. Chemother. 2013, 68, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.H.; Crosby, R.; Rasco, J.; Vaughan, D. Antimalarial activities of various 4-quinolonemethanols with special attention to WR-142,490 (mefloquine). Antimicrob. Agents Chemother. 1978, 13, 1011–1030. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Kolonoski, P.; Wu, M.; Aralar, P.A.; Inderlied, C.B.; Young, L.S. Mefloquine is active in vitro and in vivo against Mycobacterium avium complex. Antimicrob. Agents Chemother. 1999, 43, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, S.; Iso, Y.; Wan, B.; Franzblau, S.G.; Kozikowski, A.P. Design, synthesis, and SAR studies of mefloquine-based ligands as potential antituberculosis agents. ChemMedChem 2006, 1, 593–597. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernandez, I.S.; et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef] [PubMed]
- Martin-Galiano, A.J.; Gorgojo, B.; Kunin, C.M.; de la Campa, A.G. Mefloquine and new related compounds target the F(0) complex of the F(0)F(1) H(+)-ATPase of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2002, 46, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Danelishvili, L.; Wu, M.; Young, L.S.; Bermudez, L.E. Genomic approach to identifying the putative target of and mechanisms of resistance to mefloquine in mycobacteria. Antimicrob. Agents Chemother. 2005, 49, 3707–3714. [Google Scholar] [CrossRef]
- Degiacomi, G.; Chiarelli, L.R.; Recchia, D.; Petricci, E.; Gianibbi, B.; Fiscarelli, E.V.; Fattorini, L.; Manetti, F.; Pasca, M.R. The Antimalarial Mefloquine Shows Activity against Mycobacterium abscessus, Inhibiting Mycolic Acid Metabolism. Int. J. Mol. Sci. 2021, 22, 8533. [Google Scholar] [CrossRef]
- Goncalves, R.S.; Kaiser, C.R.; Lourenco, M.C.; Bezerra, F.A.; de Souza, M.V.; Wardell, J.L.; Wardell, S.M.; Henriques, M.; Costa, T. Mefloquine-oxazolidine derivatives, derived from mefloquine and arenecarbaldehydes: In vitro activity including against the multidrug-resistant tuberculosis strain T113. Bioorg. Med. Chem. 2012, 20, 243–248. [Google Scholar] [CrossRef]
- Goncalves, R.S.; Kaiser, C.R.; Lourenco, M.C.; de Souza, M.V.; Wardell, J.L.; Wardell, S.M.; da Silva, A.D. Synthesis and antitubercular activity of new mefloquine-oxazolidine derivatives. Eur. J. Med. Chem. 2010, 45, 6095–6100. [Google Scholar] [CrossRef]
- Da Silva Araujo, A.; Moraes, A.M.; Lourenco, M.C.S.; Pessoa, C.O.; da Silva, E.T.; de Souza, M.V.N. Synthesis and Antibacterial Activity of Mefloquine-Based Analogs Against Sensitive and Resistant Mycobacterium tuberculosis Strains. Curr. Top. Med. Chem. 2019, 19, 683–689. [Google Scholar] [CrossRef]
- WHO. World Health Organization Model List of Essential Medicines—22nd List, 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Frayha, G.J.; Smyth, J.D.; Gobert, J.G.; Savel, J. The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen. Pharm. 1997, 28, 273–299. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Zhu, P.J.; Hobson, J.P.; Southall, N.; Qiu, C.; Thomas, C.J.; Lu, J.; Inglese, J.; Zheng, W.; Leppla, S.H.; Bugge, T.H.; et al. Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death. Bioorg. Med. Chem. 2009, 17, 5139–5145. [Google Scholar] [CrossRef] [PubMed]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Jan, J.T.; Chen, C.M.; Hsieh, H.P.; Hwang, D.R.; Liu, H.W.; Liu, C.Y.; Huang, H.W.; Chen, S.C.; Hong, C.F.; et al. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide. Antimicrob. Agents Chemother. 2004, 48, 2693–2696. [Google Scholar] [CrossRef]
- Imperi, F.; Massai, F.; Ramachandran Pillai, C.; Longo, F.; Zennaro, E.; Rampioni, G.; Visca, P.; Leoni, L. New life for an old drug: The anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob. Agents Chemother. 2013, 57, 996–1005. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, Y. Antituberculosis activity of certain antifungal and antihelmintic drugs. Tuber. Lung Dis 1999, 79, 319–320. [Google Scholar] [CrossRef]
- Early, J.V.; Mullen, S.; Parish, T. A rapid, low pH, nutrient stress, assay to determine the bactericidal activity of compounds against non-replicating Mycobacterium tuberculosis. PLoS ONE 2019, 14, e0222970. [Google Scholar] [CrossRef]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a Drug with Many (Re)purposes. ChemMedChem 2018, 13, 1088–1091. [Google Scholar] [CrossRef]
- Fan, X.; Xu, J.; Files, M.; Cirillo, J.D.; Endsley, J.J.; Zhou, J.; Endsley, M.A. Dual activity of niclosamide to suppress replication of integrated HIV-1 and Mycobacterium tuberculosis (Beijing). Tuberculosis 2019, 116S, S28–S33. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Darby, C.M.; Rhee, K.Y.; Nathan, C. Nitazoxanide Disrupts Membrane Potential and Intrabacterial pH Homeostasis of Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2011, 2, 849–854. [Google Scholar] [CrossRef]
- Adagu, I.S.; Nolder, D.; Warhurst, D.C.; Rossignol, J.F. In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica and Trichomonas vaginalis. J. Antimicrob. Chemother. 2002, 49, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F. Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014, 110, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J. Infect. Public Health 2016, 9, 227–230. [Google Scholar] [CrossRef]
- Anastasiou, I.A.; Eleftheriadou, I.; Tentolouris, A.; Tsilingiris, D.; Tentolouris, N. In vitro Data of Current Therapies for SARS-CoV-2. Curr. Med. Chem. 2020, 27, 4542–4548. [Google Scholar] [CrossRef]
- Hoffman, P.S.; Sisson, G.; Croxen, M.A.; Welch, K.; Harman, W.D.; Cremades, N.; Morash, M.G. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductases of Helicobacter pylori, selected anaerobic bacteria and parasites, and Campylobacter jejuni. Antimicrob. Agents Chemother. 2007, 51, 868–876. [Google Scholar] [CrossRef]
- Muller, J.; Sterk, M.; Hemphill, A.; Muller, N. Characterization of Giardia lamblia WB C6 clones resistant to nitazoxanide and to metronidazole. J. Antimicrob. Chemother. 2007, 60, 280–287. [Google Scholar] [CrossRef]
- Nillius, D.; Muller, J.; Muller, N. Nitroreductase (GlNR1) increases susceptibility of Giardia lamblia and Escherichia coli to nitro drugs. J. Antimicrob. Chemother. 2011, 66, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Somvanshi, V.S.; Ellis, B.L.; Hu, Y.; Aroian, R.V. Nitazoxanide: Nematicidal mode of action and drug combination studies. Mol. Biochem. Parasitol. 2014, 193, 1–8. [Google Scholar] [CrossRef]
- Stachulski, A.V.; Taujanskas, J.; Pate, S.L.; Rajoli, R.K.R.; Aljayyoussi, G.; Pennington, S.H.; Ward, S.A.; Hong, W.D.; Biagini, G.A.; Owen, A.; et al. Therapeutic Potential of Nitazoxanide: An Appropriate Choice for Repurposing versus SARS-CoV-2? ACS Infect. Dis. 2021, 7, 1317–1331. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Lin, G.; Jiang, X.; Nathan, C. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J. Med. Chem. 2009, 52, 5789–5792. [Google Scholar] [CrossRef]
- Shigyo, K.; Ocheretina, O.; Merveille, Y.M.; Johnson, W.D.; Pape, J.W.; Nathan, C.F.; Fitzgerald, D.W. Efficacy of nitazoxanide against clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 2834–2837. [Google Scholar] [CrossRef][Green Version]
- Lam, K.K.; Zheng, X.; Forestieri, R.; Balgi, A.D.; Nodwell, M.; Vollett, S.; Anderson, H.J.; Andersen, R.J.; Av-Gay, Y.; Roberge, M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012, 8, e1002691. [Google Scholar] [CrossRef] [PubMed]
- Iacobino, A.; Giannoni, F.; Pardini, M.; Piccaro, G.; Fattorini, L. The Combination Rifampin-Nitazoxanide, but Not Rifampin-Isoniazid-Pyrazinamide-Ethambutol, Kills Dormant Mycobacterium tuberculosis in Hypoxia at Neutral pH. Antimicrob. Agents Chemother. 2019, 63, e00273-19. [Google Scholar] [CrossRef]
- Walsh, K.F.; McAulay, K.; Lee, M.H.; Vilbrun, S.C.; Mathurin, L.; Jean Francois, D.; Zimmerman, M.; Kaya, F.; Zhang, N.; Saito, K.; et al. Early Bactericidal Activity Trial of Nitazoxanide for Pulmonary Tuberculosis. Antimicrob. Agents Chemother. 2020, 64, e01956-19. [Google Scholar] [CrossRef] [PubMed]
- Upcroft, J.A.; Campbell, R.W.; Benakli, K.; Upcroft, P.; Vanelle, P. Efficacy of new 5-nitroimidazoles against metronidazole-susceptible and -resistant Giardia, Trichomonas, and Entamoeba spp. Antimicrob. Agents Chemother. 1999, 43, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Osato, T.; Umezawa, H. A new antibiotic, azomycin. J. Antibiot. 1953, 6, 182. [Google Scholar]
- Freeman, C.D.; Klutman, N.E.; Lamp, K.C. Metronidazole. A therapeutic review and update. Drugs 1997, 54, 679–708. [Google Scholar] [CrossRef]
- Wayne, L.G.; Sramek, H.A. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1994, 38, 2054–2058. [Google Scholar] [CrossRef]
- Imperiale, B.R.; Cataldi, A.A.; Morcillo, N.S. In vitro anti-tuberculosis activity of azole drugs against Mycobacterium tuberculosis clinical isolates. Rev. Argent Microbiol. 2017, 49, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Sharma, S.; Khuller, G.K. In vitro and ex vivo antimycobacterial potential of azole drugs against Mycobacterium tuberculosis H37Rv. FEMS Microbiol. Lett. 2005, 251, 19–22. [Google Scholar] [CrossRef]
- Carroll, M.W.; Jeon, D.; Mountz, J.M.; Lee, J.D.; Jeong, Y.J.; Zia, N.; Lee, M.; Lee, J.; Via, L.E.; Lee, S.; et al. Efficacy and safety of metronidazole for pulmonary multidrug-resistant tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 3903–3909. [Google Scholar] [CrossRef] [PubMed]
- Hoff, D.R.; Caraway, M.L.; Brooks, E.J.; Driver, E.R.; Ryan, G.J.; Peloquin, C.A.; Orme, I.M.; Basaraba, R.J.; Lenaerts, A.J. Metronidazole lacks antibacterial activity in guinea pigs infected with Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 4137–4140. [Google Scholar] [CrossRef][Green Version]
- Klinkenberg, L.G.; Sutherland, L.A.; Bishai, W.R.; Karakousis, P.C. Metronidazole lacks activity against Mycobacterium tuberculosis in an in vivo hypoxic granuloma model of latency. J. Infect. Dis. 2008, 198, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Furtado, L.F.V.; de Paiva Bello, A.C.P.; Rabelo, E.M.L. Benzimidazole resistance in helminths: From problem to diagnosis. Acta Trop. 2016, 162, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Knudson, S.E.; Awasthi, D.; Kumar, K.; Carreau, A.; Goullieux, L.; Lagrange, S.; Vermet, H.; Ojima, I.; Slayden, R.A. A trisubstituted benzimidazole cell division inhibitor with efficacy against Mycobacterium tuberculosis. PLoS ONE 2014, 9, e93953. [Google Scholar] [CrossRef]
- Kumar, K.; Awasthi, D.; Berger, W.T.; Tonge, P.J.; Slayden, R.A.; Ojima, I. Discovery of anti-TB agents that target the cell-division protein FtsZ. Future Med. Chem. 2010, 2, 1305–1323. [Google Scholar] [CrossRef]
- Mukherjee, T.; Boshoff, H. Nitroimidazoles for the treatment of TB: Past, present and future. Future Med. Chem. 2011, 3, 1427–1454. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lin-Hua, T.; Jantanavivat, C. Studies on a new antimalarial compound: Pyronaridine. Trans. R Soc. Trop. Med. Hyg. 1992, 86, 7–10. [Google Scholar] [CrossRef]
- Phopin, K.; Sinthupoom, N.; Treeratanapiboon, L.; Kunwittaya, S.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Antimalarial and antimicrobial activities of 8-Aminoquinoline-Uracils metal complexes. EXCLI J. 2016, 15, 144–152. [Google Scholar] [CrossRef]
- Croft, S.L.; Duparc, S.; Arbe-Barnes, S.J.; Craft, J.C.; Shin, C.S.; Fleckenstein, L.; Borghini-Fuhrer, I.; Rim, H.J. Review of pyronaridine anti-malarial properties and product characteristics. Malar. J. 2012, 11, 270. [Google Scholar] [CrossRef]
- Mori, G.; Orena, B.S.; Franch, C.; Mitchenall, L.A.; Godbole, A.A.; Rodrigues, L.; Aguilar-Perez, C.; Zemanova, J.; Huszar, S.; Forbak, M.; et al. The EU approved antimalarial pyronaridine shows antitubercular activity and synergy with rifampicin, targeting RNA polymerase. Tuberculosis 2018, 112, 98–109. [Google Scholar] [CrossRef]
- Andrade, R.M.; Chaparro, J.D.; Capparelli, E.; Reed, S.L. Auranofin is highly efficacious against Toxoplasma gondii in vitro and in an in vivo experimental model of acute toxoplasmosis. PLoS Negl. Trop. Dis. 2014, 8, e2973. [Google Scholar] [CrossRef]
- Debnath, A.; Parsonage, D.; Andrade, R.M.; He, C.; Cobo, E.R.; Hirata, K.; Chen, S.; Garcia-Rivera, G.; Orozco, E.; Martinez, M.B.; et al. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nat. Med. 2012, 18, 956–960. [Google Scholar] [CrossRef]
- Feng, L.; Pomel, S.; Latre de Late, P.; Taravaud, A.; Loiseau, P.M.; Maes, L.; Cho-Ngwa, F.; Bulman, C.A.; Fischer, C.; Sakanari, J.A.; et al. Repurposing Auranofin and Evaluation of a New Gold(I) Compound for the Search of Treatment of Human and Cattle Parasitic Diseases: From Protozoa to Helminth Infections. Molecules 2020, 25, 5075. [Google Scholar] [CrossRef] [PubMed]
- Aguinagalde, L.; Diez-Martinez, R.; Yuste, J.; Royo, I.; Gil, C.; Lasa, I.; Martin-Fontecha, M.; Marin-Ramos, N.I.; Ardanuy, C.; Linares, J.; et al. Auranofin efficacy against MDR Streptococcus pneumoniae and Staphylococcus aureus infections. J. Antimicrob. Chemother. 2015, 70, 2608–2617. [Google Scholar] [CrossRef]
- Harbut, M.B.; Vilcheze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.R., Jr.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, E.V.; Bricker-Ford, R.; Rogers, M.J.; McKerrow, J.H.; Reed, S.L. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob. Agents Chemother. 2017, 61, e01947-16. [Google Scholar] [CrossRef]
- Wallis, R.S.; Ginindza, S.; Beattie, T.; Arjun, N.; Likoti, M.; Edward, V.A.; Rassool, M.; Ahmed, K.; Fielding, K.; Ahidjo, B.A.; et al. Adjunctive host-directed therapies for pulmonary tuberculosis: A prospective, open-label, phase 2, randomised controlled trial. Lancet Respir Med. 2021, 9, 897–908. [Google Scholar] [CrossRef]
- Choi, W.H. Novel Pharmacological Activity of Artesunate and Artemisinin: Their Potential as Anti-Tubercular Agents. J. Clin. Med. 2017, 6, 30. [Google Scholar] [CrossRef]
- Patel, Y.S.; Mistry, N.; Mehra, S. Repurposing artemisinin as an anti-mycobacterial agent in synergy with rifampicin. Tuberculosis 2019, 115, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Arellanes, A.; Luna-Herrera, J.; Ruiz-Nicolas, R.; Cornejo-Garrido, J.; Tapia, A.; Yepez-Mulia, L. Antiprotozoal and antimycobacterial activities of Persea americana seeds. BMC Complement. Altern. Med. 2013, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Pérez-González, M.Z.; Gutiérrez-Rebolledo, G.A.; Yépez-Mulia, L.; Rojas-Tome, I.S.; Luna-Herrera, J.; Jiménez-Arellanes, M.A. Antiprotozoal, antimycobacterial, and anti-inflammatory evaluation of Cnidoscolus chayamansa (Mc Vaugh) extract and the isolated compounds. Biomed. Pharm. 2017, 89, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Lopez, M.; Sanchez-Hidalgo, M.; Valdivia, E.; Martinez-Bueno, M.; Maqueda, M. Are bacteriocins underexploited? Novel applications for old antimicrobials. Curr. Pharm. Biotechnol. 2011, 12, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Barrena, M.J.; Martinez-Ripoll, M.; Galvez, A.; Valdivia, E.; Maqueda, M.; Cruz, V.; Albert, A. Structure of bacteriocin AS-48: From soluble state to membrane bound state. J. Mol. Biol. 2003, 334, 541–549. [Google Scholar] [CrossRef]
- Abengozar, M.A.; Cebrian, R.; Saugar, J.M.; Garate, T.; Valdivia, E.; Martinez-Bueno, M.; Maqueda, M.; Rivas, L. Enterocin AS-48 as Evidence for the Use of Bacteriocins as New Leishmanicidal Agents. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.; Bart, J.M.; Campos-Salinas, J.; Valdivia, E.; Martinez-Bueno, M.; Gonzalez-Rey, E.; Navarro, M.; Maqueda, M.; Cebrian, R.; Perez-Victoria, J.M. Autophagic-related cell death of Trypanosoma brucei induced by bacteriocin AS-48. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 203–212. [Google Scholar] [CrossRef]
- Martin-Escolano, R.; Cebrian, R.; Maqueda, M.; Romero, D.; Rosales, M.J.; Sanchez-Moreno, M.; Marin, C. Assessing the effectiveness of AS-48 in experimental mice models of Chagas’ disease. J. Antimicrob. Chemother. 2020, 75, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Martin-Escolano, R.; Cebrian, R.; Martin-Escolano, J.; Rosales, M.J.; Maqueda, M.; Sanchez-Moreno, M.; Marin, C. Insights into Chagas treatment based on the potential of bacteriocin AS-48. Int J. Parasitol. Drugs Drug Resist. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Aguilar-Pérez, C.; Gracia, B.; Rodrigues, L.; Vitoria, A.; Cebrián, R.; Deboosere, N.; Song, O.R.; Brodin, P.; Maqueda, M.; Aínsa, J.A. Synergy between Circular Bacteriocin AS-48 and Ethambutol against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Besra, G.S. Synthesis and recycling of the mycobacterial cell envelope. Curr. Opin. Microbiol. 2021, 60, 58–65. [Google Scholar] [CrossRef] [PubMed]
- El-Saber Batiha, G.; Alqahtani, A.; Ilesanmi, O.B.; Saati, A.A.; El-Mleeh, A.; Hetta, H.F.; Magdy Beshbishy, A. Avermectin Derivatives, Pharmacokinetics, Therapeutic and Toxic Dosages, Mechanism of Action, and Their Biological Effects. Pharmaceuticals 2020, 13, 196. [Google Scholar] [CrossRef] [PubMed]
- Trunfio, M.; Scabini, S.; Mornese Pinna, S.; Rugge, W.; Alcantarini, C.; Pirriatore, V.; Di Perri, G.; Bonora, S.; Castelnuovo, B.; Calcagno, A. The Manifesto of Pharmacoenosis: Merging HIV Pharmacology into Pathocoenosis and Syndemics in Developing Countries. Microorganisms 2021, 9, 1648. [Google Scholar] [CrossRef] [PubMed]
- WHO. Soil-Transmitted Helminth Infections. Available online: https://www.who.int/news-room/fact-sheets/detail/soil-transmitted-helminth-infections (accessed on 13 October 2021).


{kind=link}
| Drug Name, Class and Structure 1 | Current Use | Rationale for TB Repurposing | Possible TB Target/ Mode of Action |
|---|---|---|---|
Avermectins (Ivermectin B1a) | Prevention of onchocerciasis and lymphatic filariasis | Active against M. tuberculosis, including M/XDR isolates (MIC = 3–6 µg/mL) | Not yet determined |
Mefloquine | Chloroquine-resistant malaria | Active against M. tuberculosis (MIC = 20–40 µM) | MmpL3 |
Niclosamide | Tapeworm infections | Active against M. tuberculosis H37R A(MIC = 0.5–1 µM) | Ionophore |
Nitazoxanide | Infections caused by Giardia intestinalis, Cryptosporidium parvum, Ascaris lumbricoides, Ancylostoma duodenale and Trichuris trichiura | Active against replicating and nonreplicating M. tuberculosis | Disruption of membrane potential and pH homeostasis. Inhibition of signaling pathways |
| Nitroimidazoles (Metronidazole)  | Helminth infections (benzimidazoles) | Active against M. tuberculosis in in vitro and ex vivo assays. Benzimidazoles show activity in murine models | FtsZ (benzimidazoles); mycolic acid biosynthesis (delamanid and pretomanid) |
Pyronaridine | Malaria | Active against M. tuberculosis in in vitro (MIC = 5 µg/mL) and ex vivo (MIC = 12.5 µg/mL) assays | Interference with nucleic acid metabolism |
Auranofin | Amebiasis | Active against M. tuberculosis (MIC = 0.5 µg/mL) | Thioredoxin reductase TrxR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezquerra-Aznárez, J.M.; Almeida da Silva, P.E.; Aínsa, J.A. Overcoming the Prokaryote/Eukaryote Barrier in Tuberculosis Treatment: A Prospect for the Repurposing and Use of Antiparasitic Drugs. Microorganisms 2021, 9, 2335. https://doi.org/10.3390/microorganisms9112335
Ezquerra-Aznárez JM, Almeida da Silva PE, Aínsa JA. Overcoming the Prokaryote/Eukaryote Barrier in Tuberculosis Treatment: A Prospect for the Repurposing and Use of Antiparasitic Drugs. Microorganisms. 2021; 9(11):2335. https://doi.org/10.3390/microorganisms9112335
Chicago/Turabian StyleEzquerra-Aznárez, José Manuel, Pedro E. Almeida da Silva, and José A. Aínsa. 2021. "Overcoming the Prokaryote/Eukaryote Barrier in Tuberculosis Treatment: A Prospect for the Repurposing and Use of Antiparasitic Drugs" Microorganisms 9, no. 11: 2335. https://doi.org/10.3390/microorganisms9112335
APA StyleEzquerra-Aznárez, J. M., Almeida da Silva, P. E., & Aínsa, J. A. (2021). Overcoming the Prokaryote/Eukaryote Barrier in Tuberculosis Treatment: A Prospect for the Repurposing and Use of Antiparasitic Drugs. Microorganisms, 9(11), 2335. https://doi.org/10.3390/microorganisms9112335