
Structure-Function Characteristics of SARS-CoV-2 Proteases and Their Potential Inhibitors from Microbial Sources



Abstract
:1. Introduction
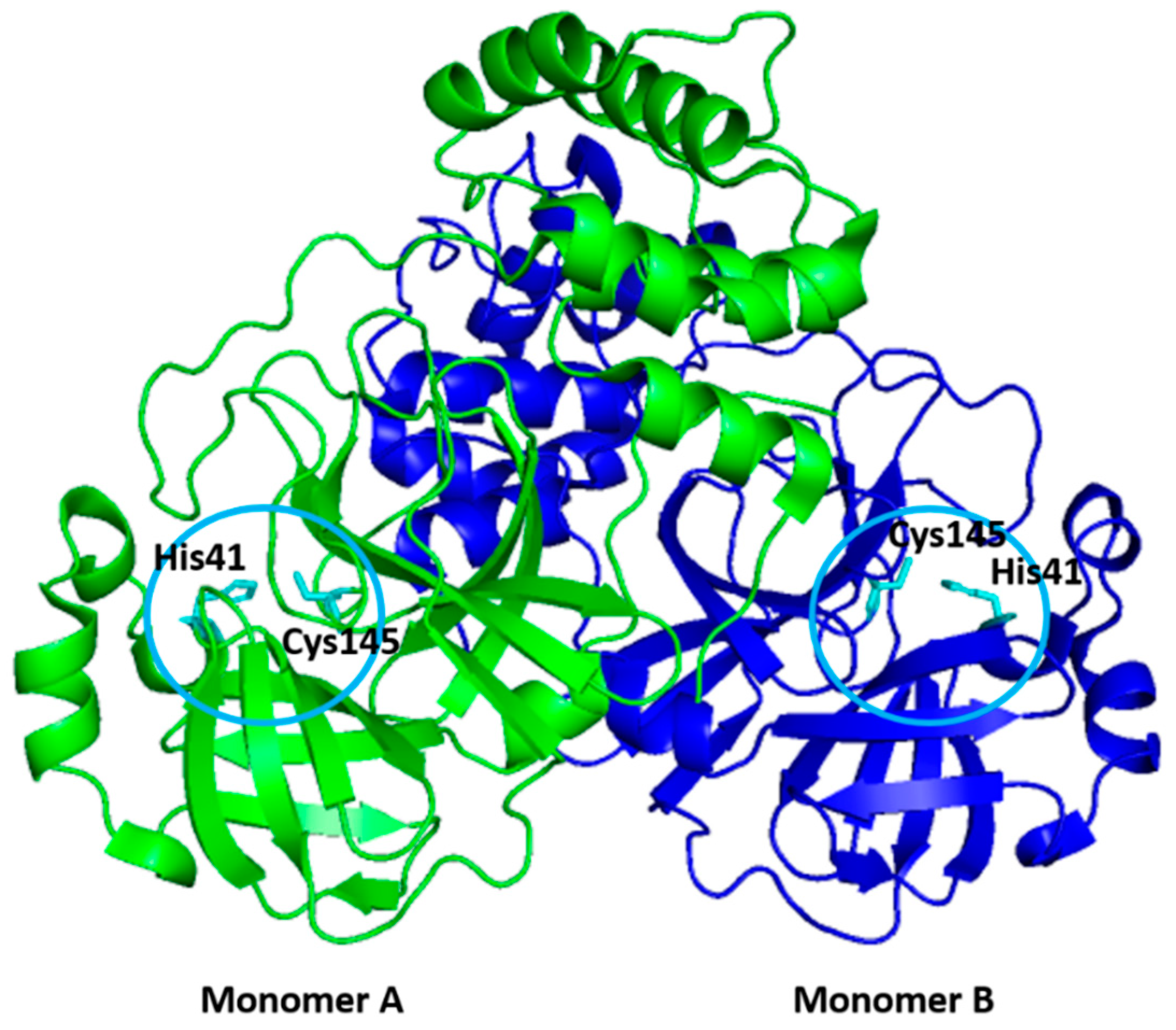
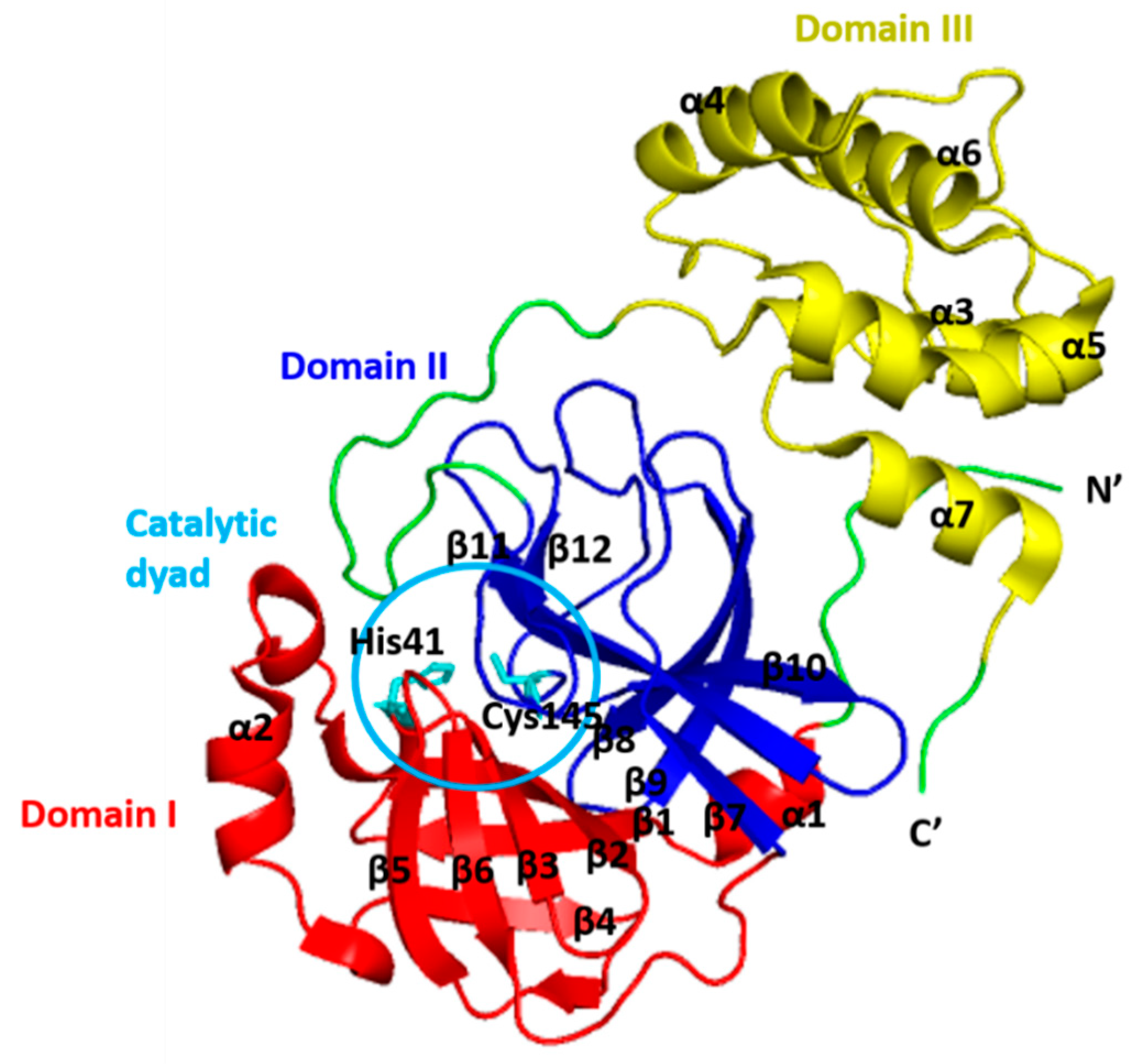
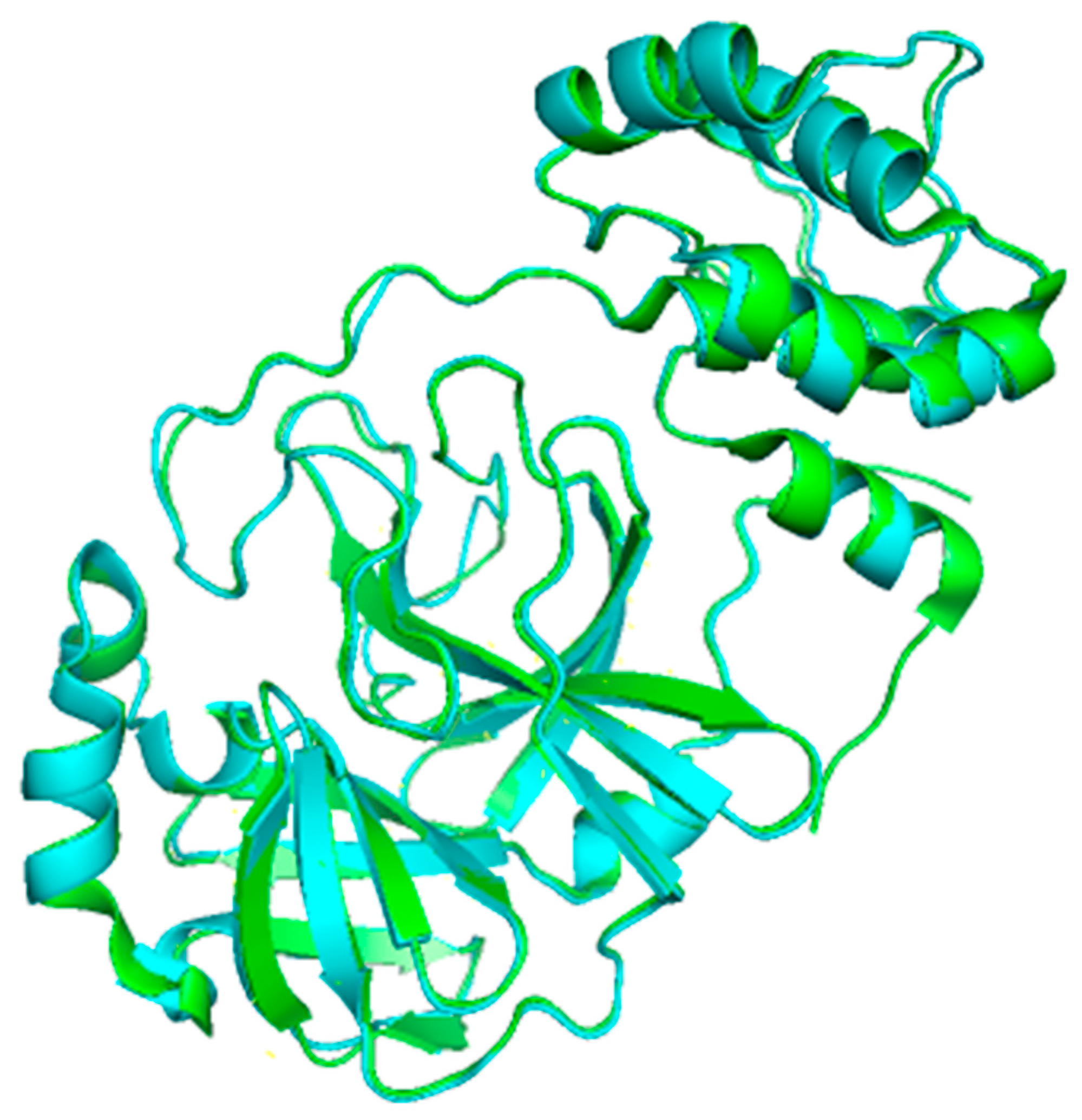
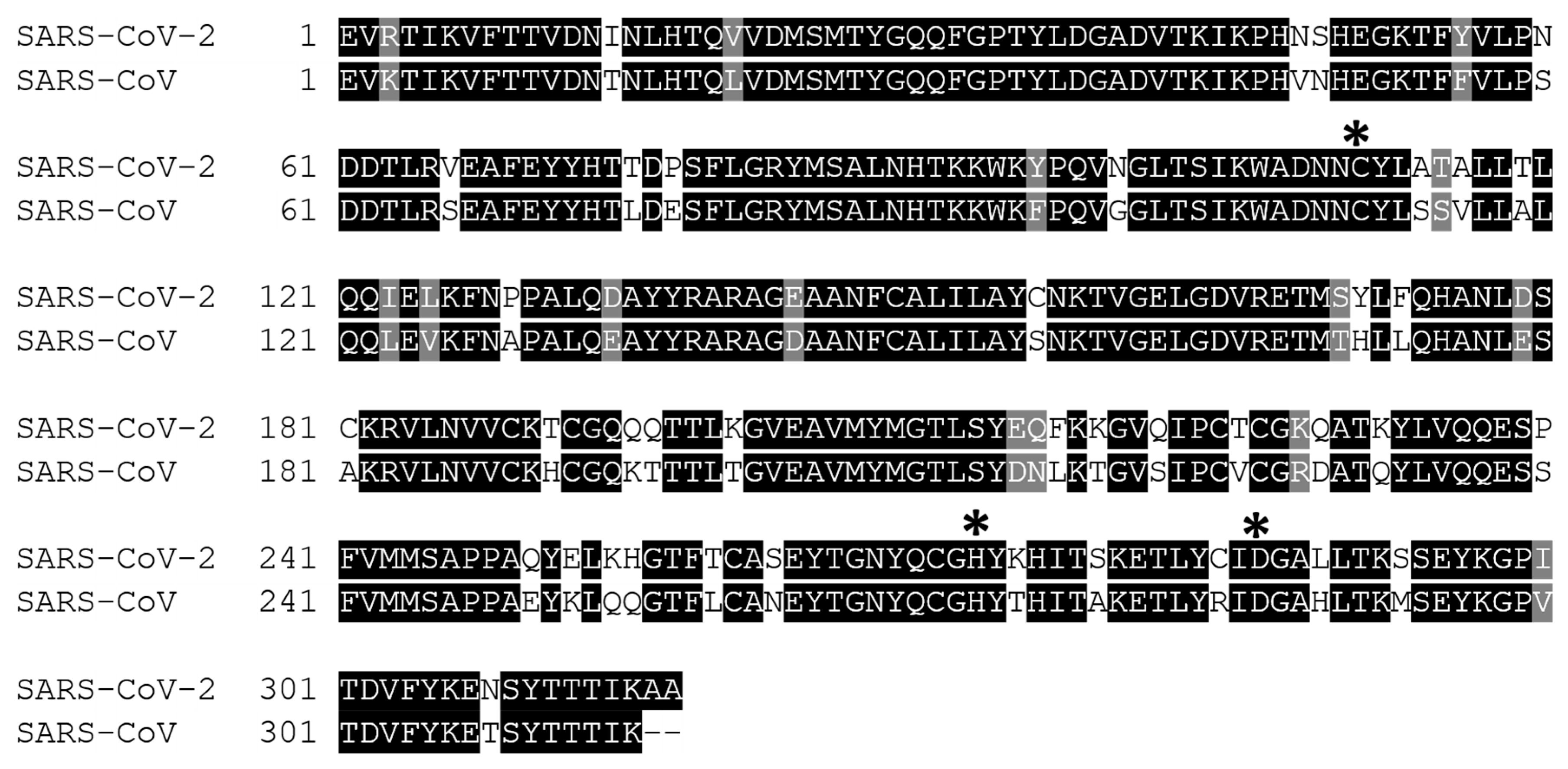
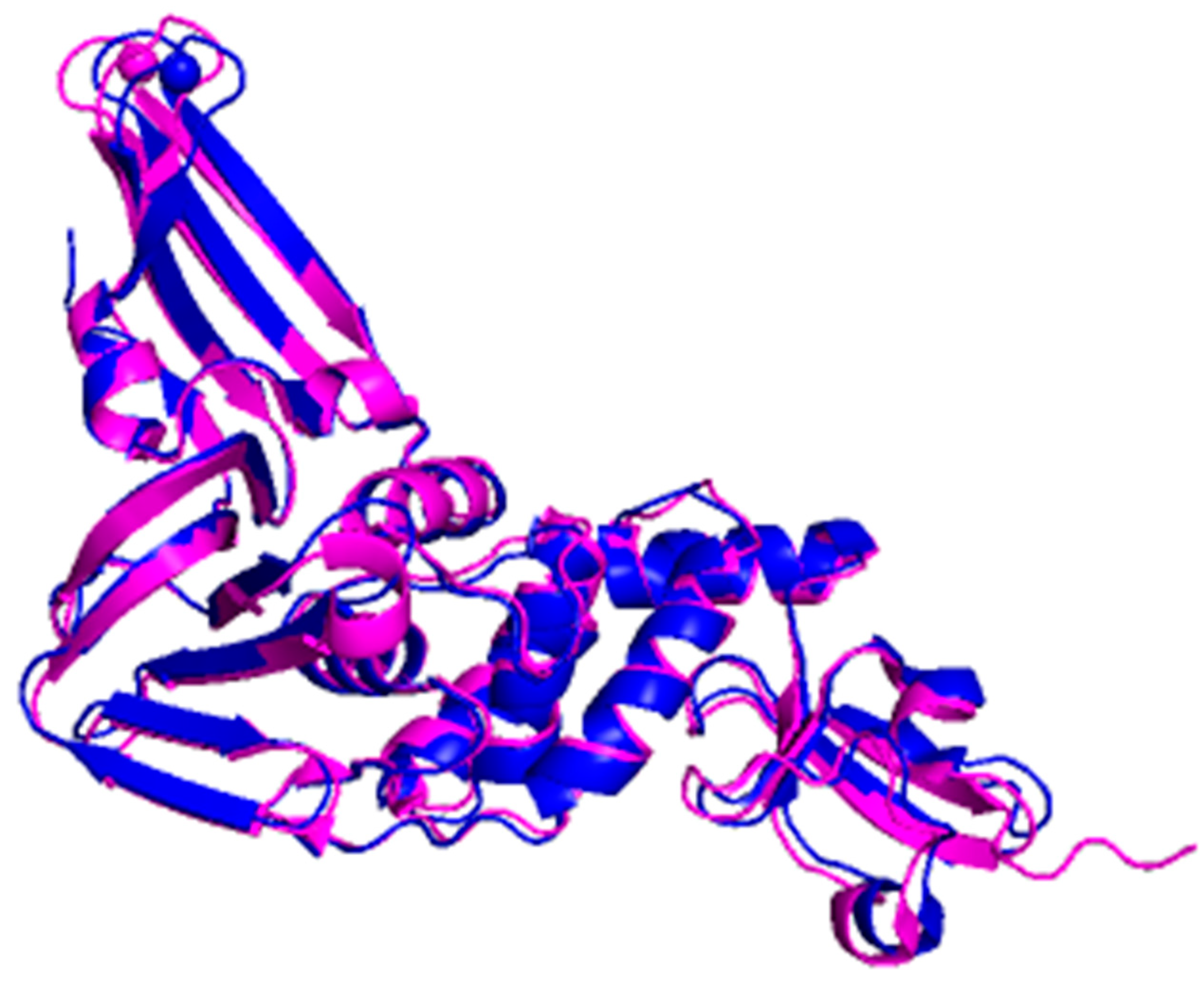
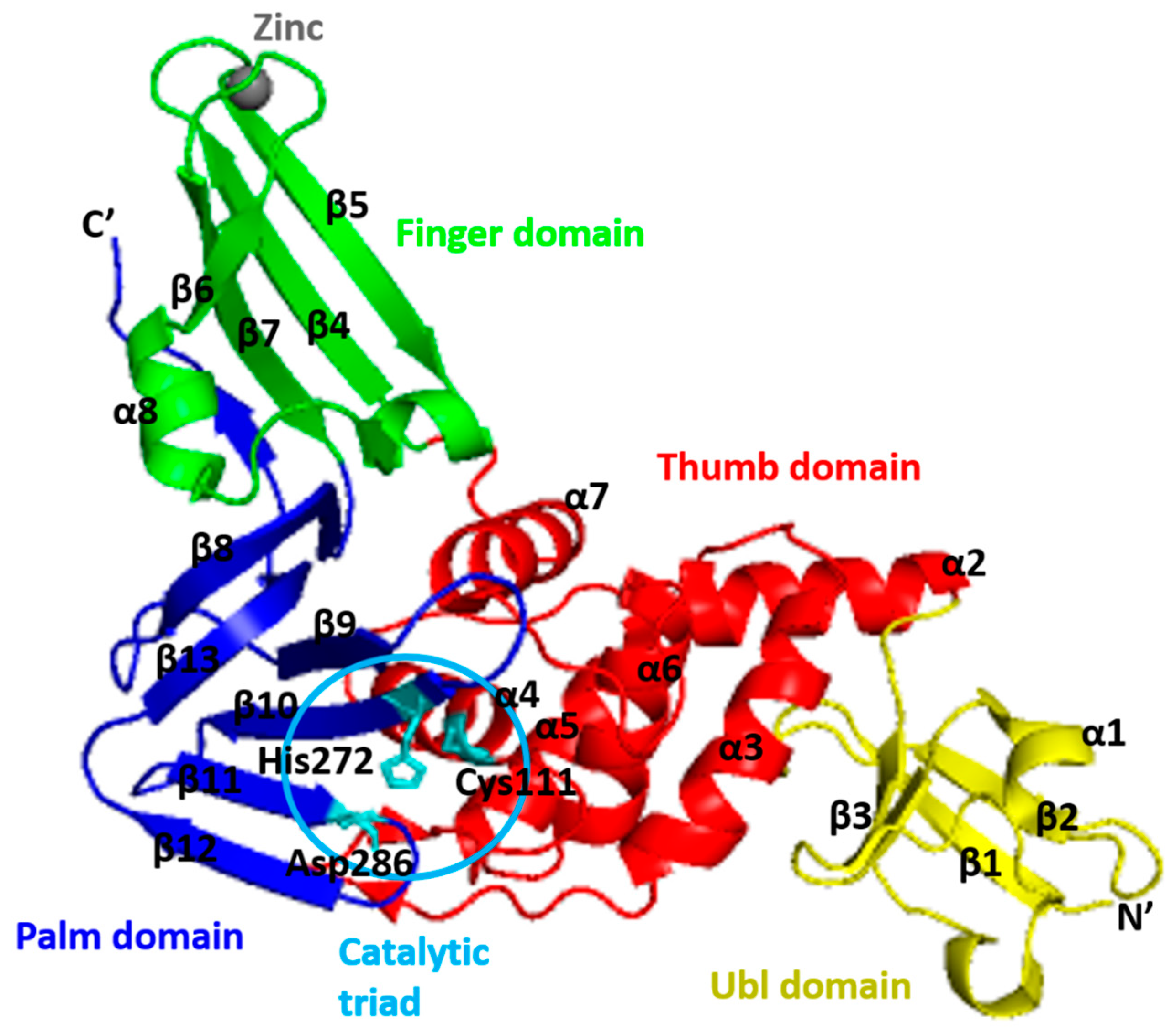
2. Structure of 3CLpro
3. Functions of 3CLpro
4. Structure of PLpro
5. Functions of PLpro
6. Microorganisms as Sources of Inhibitors Targeting SARS-CoV2 Proteases
6.1. Microbial Natural Products as Potential Inhibitor of 3CLpro-CoV2
6.2. Microbial Natural Products as a Potential Inhibitor of PLpro-CoV2
7. Possibility of Inhibitors Targeting HIV Protease for SARS-CoV2 Proteases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Haagmans, B.L.; Sola, I. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-ncov and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Goyal, B.; Goyal, D. Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [PubMed] [Green Version]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Van Regenmortel, M.H.V.; Fauquet, C.M.; Bishop, D.H.L.; Carstens, E.B.; Estes, M.K.; Lemon, S.M.; Maniloff, J.; Mayo, M.A.; McGeoch, D.J.; Pringle, C.R.; et al. Virus Taxonomy: Classification and Nomenclature of Viruses Seventh Report of the International Committee on Taxonomy of Viruses; Academic Press: San Diego, CA, USA, 2000; pp. 835–849. [Google Scholar]
- Malik, Y.A. Properties of coronavirus and SARS-CoV-2. Malays. J. Pathol. 2020, 42, 3–11. [Google Scholar] [PubMed]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease 2019 (COVID-19) Situation Report–89. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ (accessed on 18 April 2020).
- Cui, J.; Li, F.; Shi, Z. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Alfaraj, S.H.; Al-Tawfiq, J.A.; Assiri, A.Y.; Alzahrani, N.A.; Alanazi, A.A.; Memish, Z.A. Clinical predictors of mortality of middle east respiratory syndrome coronavirus (MERS-CoV) infection: A cohort study. Travel Med. Infect. Dis. 2019, 29, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Osipiuk, J.; Azizi, S.A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Hussain, S.; Pan, J.; Chen, Y.; Yang, Y.; Xu, J.; Peng, Y.; Wu, Y.; Li, Z.; Zhu, Y.; Tien, P.; et al. Identification of novel subgenomic rnas and noncanonical transcription initiation signals of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 5288–5295. [Google Scholar] [CrossRef] [Green Version]
- Czub, M.; Weingartl, H.; Czub, S.; He, R.; Cao, J. Evaluation of modified vaccinia virus ankara based recombinant SARS vaccine in ferrets. Vaccine 2005, 23, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Ramajayam, R.; Tan, K.; Liang, P. Recent Development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011, 39, 1371–1375. [Google Scholar] [CrossRef]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus Hcov-EMC also has an "Achilles’ Heel": Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248–250. [Google Scholar] [CrossRef]
- van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 2012, 3, e00473-12. [Google Scholar] [CrossRef] [Green Version]
- De Diego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in Vitro and in Vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [Green Version]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.; Masters, P.S. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 2003, 77, 4597–4608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortego, J.; Ceriani, J.E.; Patiño, C.; Plana, J.; Enjuanes, L. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, Y.; Teoh, K.; Lo, J.; Chan, C.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef]
- Kuo, C.J.; Chi, Y.H.; Hsu, J.T.A.; Liang, P.H. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 2004, 318, 862–867. [Google Scholar] [CrossRef]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.C.; Chang, H.C.; Chou, C.Y.; Tsai, P.J.; Lin, P.I.; Chang, G.G. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005, 280, 22741–22748. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.Y.; Chang, W.H.; Liang, J.Y.; Lin, L.L.; Chang, G.G.; Chang, H.P. Essential covalent linkage between the chymotrypsin-like domain and the extra domain of the SARS-CoV main protease. J. Biochem. 2010, 148, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Kang, X. Activation and maturation of SARS-CoV main protease. Protein Cell 2011, 2, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J. Gen. Virol. 2021, 102, 001558. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Comm. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Li, S.; Xue, F.; Zou, Y.; Chen, C.; Bartlam, M.; Rao, Z. Structure of the main protease from a global infectious human coronavirus, HCoV-HKU1. J. Virol. 2008, 82, 8647–8655. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Zhou, F.; Huang, Z.; Ma, X.; Natarajan, K.; Zhang, M.; Huang, Y.; Su, H. Molecular insights into small molecule drug discovery for SARS-CoV-2. Angew. Chem. 2020, 133, 9873–9886. [Google Scholar] [CrossRef]
- Shi, J.; Wei, Z.; Song, J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: Defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004, 279, 24765–24773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.Y.; Chang, H.C.; Hsu, W.C.; Lin, T.Z.; Lin, C.H.; Chang, G.G. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry 2004, 43, 14958–14970. [Google Scholar] [CrossRef] [PubMed]
- Barrila, J.; Bacha, U.; Freire, E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 2006, 45, 14908–14916. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhang, J.; Hu, T.; Chen, K.; Jiang, H.; Shen, X. Residues on the dimer interface of SARS coronavirus 3C-like protease: Dimer stability characterization and enzyme catalytic activity analysis. J. Biochem. 2008, 143, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Hu, T.; Zhang, J.; Chen, J.; Chen, K.; Ding, J.; Jiang, H.; Shen, X. Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: Crystal structure with molecular dynamics simulations. J. Biol. Chem. 2008, 283, 554–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.Y.; Chou, C.Y.; Chang, H.C.; Hsu, W.C.; Chang, G.G. Correlation between dissociation and catalysis of SARS-CoV main protease. Arch. Biochem. Biophys. 2008, 472, 34–42. [Google Scholar] [CrossRef]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Zhang, Y.; Li, L.; Wang, K.; Chen, S.; Chen, J.; Ding, J.; Jiang, H.; Shen, X. Two adjacent mutations on the dimer interface of SARS coronavirus 3C-like protease cause different conformational changes in crystal structure. Virology 2009, 388, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrila, J.; Gabelli, S.B.; Bacha, U.; Amzel, L.M.; Freire, E. Mutation of Asn28 disrupts the dimerization and enzymatic activity of SARS 3CLpro. Biochemistry 2010, 49, 4308–4317. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Gao, F. An overview of potential inhibitors targeting non-structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 4868–4883. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Gorbalenya, A.E.; Snijder, E.J. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef] [PubMed]
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Mesecar, A.D.; Denison, M.R. Chimeric exchange of coronavirus NSP5 proteases (3CLpro) identifies common and divergent regulatory determinants of protease activity. J. Virol. 2013, 87, 12611–12618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Lu, Y.; Denison, M.R. Intracellular and in vitro-translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology 1996, 222, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanjanahaluethai, A.; Baker, S.C. Identification of mouse hepatitis virus papain-like proteinase 2 activity. J. Virol. 2000, 74, 7911–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, E.F.; Graham, R.L.; Sims, A.C.; Denison, M.R.; Baric, R.S. Analysis of murine hepatitis virus strain A59 temperature-sensitive mutant TS-LA6 suggests that nsp10 plays a critical role in polyprotein processing. J. Virol. 2007, 81, 7086–7098. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.F.; Kuo, C.J.; Chang, K.T.; Chang, H.C.; Chou, C.C.; Ko, T.P.; Shr, H.L.; Chang, G.G.; Wang, A.H.J.; Liang, P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [Green Version]
- Tomar, S.; Johnston, M.L.; John, S.E.S.; Osswald, H.L.; Nyalapatla, P.R.; Paul, L.N.; Ghosh, A.K.; Denison, M.R.; Mesecar, A.D. Ligand-Induced dimerization of middle east respiratory syndrome (MERS) coronavirus NSP5 protease (3CLpro) implications for NSP5 regulation and the development of antivirals. J. Biol. Chem. 2015, 290, 19403–19422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Qi, Y.; Teng, X.; Yang, Z.; Wei, P.; Zhang, C.; Tan, L.; Zhou, L.; Liu, Y.; Lai, L. Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C-like proteinase. J. Biol. Chem. 2010, 285, 28134–28140. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Jonas, F.; Shen, C.; Higenfeld, R.; Higenfeld, R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 2010, 1, 59–74. [Google Scholar] [CrossRef]
- Krichel, B.; Falke, S.; Hilgenfeld, R.; Redecke, L.; Uetrecht, C. Processing of the SARS-CoV pp1a/ab nsp7–10 region. Biochem. J. 2020, 477, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deming, D.J.; Graham, R.L.; Denison, M.R.; Baric, R.S. Processing of open reading frame 1A replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J. Virol. 2007, 81, 10280–10291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stobart, C.C.; Lee, A.S.; Lu, X.; Denison, M.R. Temperature-sensitive mutants and revertants in the coronavirus nonstructural protein 5 protease (3CLpro) define residues involved in long-distance communication and regulation of protease activity. J. Virol. 2012, 86, 4801–4810. [Google Scholar] [CrossRef] [Green Version]
- Stokes, H.L.; Baliji, S.; Hui, C.G.; Sawicki, S.G.; Baker, S.C.; Siddell, S.G. A new cistron in the murine hepatitis virus replicase gene. J. Virol. 2010, 84, 10148–10158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosken, Y.K.; Cholko, T.; Lou, Y.C.; Wu, K.P.; Chang, C.E.A. Insights into dynamics of inhibitor and ubiquitin-like protein binding in SARS-CoV-2 papain-like protease. Front. Mol. Biosci. 2020, 7, 174. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Ismail, M.I.; Bauer, M.R.; Bekhit, A.A.; Boeckler, F.M. Supporting SARS-CoV-2 papain-like protease drug discovery: In silico methods and benchmarking. Front. Chem. 2020, 8, 592289. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Ratia, K.; Saikatendu, K.S.; Santarsiero, B.D.; Barretto, N.; Baker, S.C.; Stevens, R.C.; Mesecar, A.D. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [Google Scholar] [CrossRef] [Green Version]
- Clasman, J.R.; Báez-Santos, Y.M.; Mettelman, R.C.; O’Brien, A.; Baker, S.C.; Mesecar, A.D. X-ray structure and enzymatic activity profile of a core papain-like protease of MERS coronavirus with utility for structure-based drug design. Sci. Rep. 2017, 7, 40292. [Google Scholar] [CrossRef] [Green Version]
- Henderson, J.A.; Verma, N.; Harris, R.C.; Liu, R.; Shen, J. Assessment of proton-coupled conformational dynamics of SARS and MERS coronaviruses papain-like proteases: Implication for designing broad-spectrum antiviral inhibitors. Chem. Phys. 2020, 153, 115101. [Google Scholar]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Chou, Y.W.; Cheng, S.C.; Lai, H.Y.; Chou, C.Y. Differential domain structure stability of the severe acute respiratory syndrome coronavirus papain-like protease. Arch. Biochem. Biophys. 2012, 520, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Báez-Santos, Y.M.; Barraza, S.J.; Wilson, M.W.; Agius, M.P.; Mielech, A.M.; Davis, N.M.; Baker, S.C.; Larsen, S.D.; Mesecar, A.D. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014, 57, 2393–2412. [Google Scholar] [CrossRef] [Green Version]
- Báez-Santos, Y.M.; John, S.E.S.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiel, I.A.; De Vries, L.A.; Reggiori, F.; Rottier, P.J.; De Haan, C.A. Dynamics of coronavirus replication transcription complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef] [Green Version]
- Oostra, M.; Hagemeijer, M.C.; Van Gent, M.; Bekker, C.P.; Te Lintelo, E.G.; Rottier, P.J.; De Haan, C.A. Topology and membrane anchoring of the coronavirus replication complex: Not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 2008, 82, 12392–12405. [Google Scholar] [CrossRef] [Green Version]
- Kandeel, M.; Abdelrahman, A.H.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.N.; Morsy, M.A.; Ibrahim, M.A. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J. Biomol. Struc. Dyn. 2020, 39, 1–8. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Maiti, B.K. Can papain-like protease inhibitors halt SARS-CoV-2 replication? ACS Pharmacol. Transl. Sci. 2020, 3, 1017–1019. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.K.; Baker, S.C.; Li, K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [Green Version]
- Ratia, K.; Kilianski, A.; Báez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the ubiquitin-linkage specificity and deisgylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, B.; Leach, C.A.; Goldenberg, S.J.; Francis, D.M.; Kodrasov, M.P.; Tian, X.; Shanks, J.; Sterner, D.E.; Bernal, A.; Mattern, M.R.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008, 17, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Dzimianski, J.V.; Scholte, F.E.; Bergeron, É.; Pegan, S.D. ISG15: It’s complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef] [PubMed]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Sulea, T.; Lindner, H.A.; Purisima, E.O.; Menard, R. Deubiquitination, a new function of the severe acute respiratory syndrome coronavirus papain-like protease? J. Virol. 2005, 79, 4550–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Békés, M.; Ekkebus, R.; Ovaa, H.; Huang, T.T.; Lima, C.D. Recognition of Lys48-linked di-ubiquitin and deubiquitinating activities of the SARS coronavirus papain-like protease. Mol. Cell 2016, 62, 572–585. [Google Scholar] [CrossRef] [Green Version]
- Clemente, V.; D’Arcy, P.; Bazzaro, M. Deubiquitinating enzymes in coronaviruses and possible therapeutic opportunities for COVID-19. Int. J. Mol. Sci. 2020, 21, 3492. [Google Scholar] [CrossRef]
- McClain, C.B.; Vabret, N. SARS-CoV-2: The many pros of targeting PLpro. Signal Transduct. Target Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Demain, A.L. Antibiotics: Natural products essential for human health. Med. Res. Rev. 2009, 29, 821–841. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Knauf, C. How gut microbes talk to organs: The role of endocrine and nervous routes. Mol. Met. 2016, 5, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Singh, B.K.; MacDonald, C.A. Drug discovery from uncultivable microorganisms. Drug Discov. Today 2010, 15, 792–799. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Tripathi, A.; Misra, K. Molecular docking: A structure-based drug designing approach. JSM Chem. 2017, 5, 1042–1047. [Google Scholar]
- Wang, Z.; Jia, J.; Wang, L.; Li, F.; Wang, Y.; Jiang, Y.; Song, X.; Qin, S.; Zheng, K.; Ye, J.; et al. Anti-HSV-1 activity of Aspergillipeptide D, a cyclic pentapeptide isolated from fungus Aspergillus sp. SCSIO 41501. Virol. J. 2020, 17, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhu, T.; Gu, Q.; Xi, R.; Wang, W.; Li, D. Structures and antiviral activities of butyrolactone derivatives isolated from Aspergillus terreus MXH-23. Ocean Univ. China 2014, 13, 1067–1070. [Google Scholar] [CrossRef]
- Ma, X.; Nong, X.; Ren, Z.; Wang, J.; Liang, X.; Wang, L.; Xi, S. Antiviral peptides from marine gorgonian-derived fungus Aspergillus sp. SCSIO 41501. Tetrahedron Lett. 2017, 58, 1151–1155. [Google Scholar] [CrossRef]
- Houda Sara, N.; Rachid, B.; Julio, G.; Jesus, R.-S.M.; Teresa, V. In vitro antimicrobial, antiviral and cytotoxicity activities of Aspergillus oryzae isolated from El-Baida Marsh in Algeria. J. Drug Deliv. Ther. 2020, 10, 191–195. [Google Scholar] [CrossRef]
- Koehler, P.; Bassetti, M.; Chakrabarti, A.; Chen, S.C.A.; Colombo, A.L.; Hoenigl, M.; Klimko, N.; Lass-Flörl, C.; Oladele, R.O.; Vinh, D.C.; et al. Defining and managing COVID-19-associated pulmonary aspergillosis: The 2020 ECMM/ISHAM consensus criteria for research and clinical guidance. Lancet Infect. Dis. 2021, 21, E149–E162. [Google Scholar] [CrossRef]
- Zacharof, M.P.; Lovitt, R.W. Bacteriocins produced by lactic acid bacteria a review article. APCBEE Procedia 2012, 2, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Abdelmohsen, U.R.; Rateb, M.E. Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef]
- Rao, P.; Shukla, A.; Parmar, P.; Rawal, R.M.; Patel, B.; Saraf, M.; Goswami, D. Reckoning a fungal metabolite, pyranonigrin A as a potential main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamic simulation. Biophys. Chem. 2020, 264, 106425. [Google Scholar] [CrossRef]
- Quimque, M.T.J.; Notarte, K.I.R.; Fernandez, R.A.T.; Mendoza, M.A.O.; Liman, R.A.D.; Lim, J.A.K.; Pilapil, L.A.E.; Ong, J.K.H.; Pastrana, A.M.; Khan, A.; et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2021, 39, 4316–4333. [Google Scholar] [CrossRef]
- El-Hawary, S.S.; Mohammed, R.; Bahr, H.S.; Attia, E.Z.; El-Katatny, M.H.; Abelyan, N.; Al-Sanea, M.M.; Moawad, A.S.; Abdelmohsen, U.R. Soybean-associated endophytic fungi as potential source for anti-COVID-19 metabolites supported by docking analysis. J. Appl. Microbiol. 2021, 131, 1193–1211. [Google Scholar] [CrossRef]
- Prajapati, J.; Patel, R.; Goswami, D.; Saraf, M.; Rawal, R.M. Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Comput. Biol. Med. 2021, 135, 104568. [Google Scholar] [CrossRef]
- Alam, S.; Sadiqi, S.; Sabir, M.; Nisa, S.; Ahmad, S.; Abbasi, S.W. Bacillus species; a potential source of anti-SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef]
- Naidoo, D.; Roy, A.; Kar, P.; Mutanda, T.; Anandraj, A. Cyanobacterial metabolites as promising drug leads against the Mpro and PLpro of SARS-CoV-2: An in silico analysis. J. Biomol. Struct. Dyn. 2021, 16, 6218–6230. [Google Scholar] [CrossRef]
- Balmeh, N.; Mahmoudi, S.; Fard, N.A. Manipulated bio antimicrobial peptides from probiotic bacteria as proposed drugs for COVID-19 disease. Inform. Med. Unlocked. 2021, 23, 100515. [Google Scholar] [CrossRef]
- Rao, P.; Patel, R.; Shukla, A.; Parmar, P.; Rawal, R.M.; Saraf, M.; Goswami, D. Identifying structural-functional analogue of GRL0617, the only well-established inhibitor for papain-like protease (PLpro) of SARS-CoV-2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol. Divers. 2021, 6, 1–21. [Google Scholar]
- Bansal, P.; Kumar, R.; Singh, J.; Dhanda, S. In silico molecular docking of SARS-CoV-2 surface proteins with microbial non-ribosomal peptides: Identification of potential drugs. J. Proteins Proteom. 2021, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020, 39, 3396–3408. [Google Scholar] [CrossRef]
- Sharma, A.; Goyal, S.; Yadav, A.K.; Kumar, P.; Gupta, L. In-silico screening of plant-derived antivirals against main protease, 3CLpro and endoribonuclease, NSP15 proteins of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.E.; Amen, Y.; Ashour, A.; Assaf, H.K.; Hassan, H.A.; Abdel-Rahman, I.M.; Sayed, A.M.; Shimizu, K. In silico study of natural compounds from sesame against COVID-19 by targeting Mpro, PLpro and RdRp. RSC Adv. 2021, 11, 22398. [Google Scholar] [CrossRef]
- Contreras-Puentes, N.; Alviz-Amador, A. Virtual screening of natural metabolites and antiviral drugs with potential inhibitory activity against 3CL-PRO and PL-PRO. Biomed. Pharmacol. 2020, 13, 933–941. [Google Scholar] [CrossRef]
- Van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castaño-Espriu, L.; Chang, C.; Clark, T.N.; et al. The natural products atlas: An open access knowledge base for microbial natural products discovery. ACS Central Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef] [Green Version]
- El-Neketi, M.; Ebrahim, W.; Lin, W.; Gedara, S.; Badria, F.; Saad, H.E.A.; Lai, D.; Proksch, P. Alkaloids and polyketides from Penicillium citrinum, an endophyte isolated from the moroccan plant ceratonia siliqua. J. Nat. Prod. 2013, 76, 1099–1104. [Google Scholar] [CrossRef]
- Riko, R.; Nakamura, H.; Shindo, K. Studies on pyranonigrins-isolation of pyranonigrin E and biosynthetic studies on pyranonigrin A. J. Antibiot. 2014, 67, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Nong, X.H.; Wang, Y.F.; Zhang, X.Y.; Zhou, M.P.; Xu, X.Y.; Qi, S.H. Territrem and butyrolactone derivatives from a marine-derived fungus Aspergillus terreus. Mar. Drugs 2014, 12, 6113–6124. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.T.; Qi, Q.Y.; Ma, K.; Pei, Y.F.; Han, J.J.; Xu, W.; Li, E.W.; Liu, H.W. Depside α -glucosidase inhibitors from a culture of the mushroom Stereum hirsutum. Planta Med. 2014, 80, 918–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elnagdy, S.; AlKhazindar, M. The potential of antimicrobial peptides as an antiviral therapy against COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Maleki, M.S.M.; Rostamian, M.; Madanchi, H. Antimicrobial peptides and other peptide-like therapeutics as promising candidates to combat SARS-CoV-2. ERATCK 2021, 19, 1205–1217. [Google Scholar]
- Verma, Y.K.; Verma, R.; Tyagi, N.; Behl, A.; Kumar, S.; Gangenahalli, G.U. COVID-19 and its therapeutics: Special emphasis on mesenchymal stem cells based therapy. Stem Cell Rev. Rep. 2021, 17, 113–131. [Google Scholar] [CrossRef]
- Galmarini, O.L.; Stodola, F.H.; Raper, K.B.; Fennell, D.I. Fonsecin, a naphthopyrone pigment from a mutant of Aspergillus fonsecaeus. Nature 1962, 195, 502–503. [Google Scholar] [CrossRef]
- Huang, L.H.; Xu, M.Y.; Li, H.J.; Li, J.Q.; Chen, Y.X.; Ma, W.Z.; Li, Y.P.; Xu, J.; Yang, D.P.; Lan, W.J. Amino Acid-directed strategy for inducing the marine-derived fungus Scedosporium apiospermum F41-1 to maximize alkaloid diversity. Org. Lett. 2017, 19, 4888–4891. [Google Scholar] [CrossRef]
- Peng, J.; Lin, T.; Wang, W.; Xin, Z.; Zhu, T.; Gu, Q.; Li, D. Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013, 76, 1133–1140. [Google Scholar] [CrossRef]
- Adeoye, A.O.; Oso, B.J.; Olaoye, I.F.; Tijjani, H.; Adebayo, A.I. Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. J. Biomol. Struct. Dyn. 2020, 39, 3469–3479. [Google Scholar] [CrossRef] [PubMed]
- Loll, P.J.; Upton, E.C.; Nahoum, V.; Economou, N.J.; Cocklin, S. The high resolution structure of tyrocidine A reveals an amphipathic dimer. Biochim. Biophys. Acta. 2014, 1838, 1199–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, A.; Pierson, D.; Mishra, S.; Koenig, D.W.; Demain, A.L. Gramicidin S production by Bacillus brevis in Simulated Microgravity. Curr. Microbiol. 1997, 34, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar]
- Wang, Z.; Chen, X.; Lu, Y.; Chen, F.; Zhang, W. Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci. Trends 2020, 14, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Jeon, S.; Shin, H.Y.; Kim, M.J.; Seong, Y.M.; Lee, W.J.; Choe, K.W.; Kang, Y.M.; Lee, B.; Park, S.J. Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: The application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by Quantitative RT-PCR. J. Korean Med. Sci. 2020, 35, e79. [Google Scholar] [CrossRef]
- Tran, L.; Tam, D.N.H.; Elhadad, H.; Hien, N.M.; Huy, N.T. Evaluation of COVID-19 protease and HIV inhibitors interactions. Acta Pharm. 2021, 72, 1–8. [Google Scholar] [CrossRef]
- Raphael, V.P.; Shanmughan, S.K. Computational evaluation of the inhibition efficacies of HIV antivirals on SARS-CoV-2 (COVID-19) protease and identification of 3D pharmacophore and hit compounds. Adv. Pharmacol. Sci. 2020, 2020, 8818008. [Google Scholar] [CrossRef]
- Bolcato, G.; Bissaro, M.; Pavan, M.; Sturlese, M.; Moro, S. Targeting the coronavirus SARS-CoV-2: Computational insights into the mechanism of action of the protease inhibitors lopinavir, ritonavir and nelfinavir. Sci. Rep. 2020, 10, 20927. [Google Scholar] [CrossRef]
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tozser, J. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2′s main protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef]
- Razali, R.; Subbiah, V.K.; Budiman, C. Technical data of heterologous expression and purification of SARS-CoV-2 proteases using Escherichia coli system. Data 2021, 6, 99. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound/Peptide | Type | Organism | Ref. |
|---|---|---|---|
| Potential Inhibitor for 3CLpro-CoV2 | |||
| Citriquinochroman | Hydroquinolones | Penicillium citrinum | [108] |
| Holyrine B | Indolocarbazole | Streptococcus mutans | |
| Proximicin C | Furan analogue of netropsin | Verrucosispora maris | |
| Pityriacitrin B | Ultraviolet-absorbing indole alkaloid | Malassezia furfur | |
| (+)-Anthrabenzoxocinone | Aromatic polyketide | Streptomyces sp. | |
| Penimethavone A | Flavone | Penicillium chrysogenum | |
| Pyranonigrin A | Pyranopyrroles | Aspergillus niger | [109] |
| Altertoxin V | Perylene quinone | Alternaria spp. | [110] |
| Altertoxin II | Perylene quinone | Alternaria spp. | |
| Penicillixanthone A | Xanthone dimer | Aspergillus fumigates | |
| Cytochalasin Z8 | Cytochalasan alkaloid | Spicaria elegans | |
| Chloropupukeanolide A | Spiroketal peroxides | Pestalotiopsis fici | |
| Phomasetin | Alkaloids | Phoma sp. | |
| Isochaetochromin D1 | Polyketide | Fusarium sp. | |
| Aspergilol H (R) | Anthraquinones | Aspergillus versicolor | |
| Aspergilol H (S) | Anthraquinones | Aspergillus versicolor | |
| 11a-Dehydroxyiso-terreulactone A | Terpenoid | Aspergillus terreus SCSGAF0162 | |
| Arisugacin A | Aromatic ether organic heterotetracyclic | Penicillium sp. | |
| Aspernolide A | Butyrolactone secondary metabolite | Cladosporium cladosporioides | |
| Rhodatin | Meroterpenoid | Rhodotus palmatus | |
| Scedapin C | Fumiquinazoline alkaloids | Scedosporium apiospermum | |
| Scequinadoline A | Fumiquinozalines | Dichotomomyces cejpii | |
| 14S-Oxoglyantrypine | Indole alkaloids containing pyrazinoquinazoline-derivative framework | Cladosporium sp. PJX-41 | |
| Deoxynortryptoquivaline | Alkaloid | Cladosporium sp. PJX-41 | |
| Quinadoline B | Alkaloid | Cladosporium sp. PJX-41 | |
| Norquinadoline A | Fumiquinazoline alkaloids | Cladosporium sp. PJX-41 | |
| Asperterrestide A (S) | Cyclic tetrapeptide | Aspergillus terreus SCSGAF0162 | |
| Asperterrestide A (R) | Cyclic tetrapeptide | Aspergillus terreus SCSGAF0162 | |
| Rubrolide S | Rubrolide | Aspergillus terreus OUCMDZ-1925 | |
| Isoaspulvinone | Aspulvinone | Aspergillus terreus | |
| Aspergilide B1 | Butenolide | Aspergillus ostianus strain 01F313 | [111] |
| 3α-hydroxy-3,5-dihydromonacolin L | Polyketide | Aspergillus terreus | |
| 2-cyclohexyl-∼{N}-pyridin-3-yl-ethanamide | Non-polymer | Aspergillus terreus | |
| Sulochrin | Benzophenone | Aspergillus terreus, Aureobasidium | |
| Emodin | Trihydroxyanthraquinone | Aspergillus ochraceus, Aspergillus wentii, Aspergillus terreus | |
| Reticulol (6-demethylkigelin) | Isocoumarin | Aspergillus terreus | |
| Aspergiketal | Spiroketal | Aspergillus terreus | |
| Terrelactone A | Butyrolactone | Aspergillus terreus | |
| Dihydrocitrinone | Isocoumarin | Aspergillus terreus | |
| 4-Hydroxykigelin | Isocoumarin | Aspergillus terreus | |
| Terreic acid | Quinone epoxide | Aspergillus terreus | |
| Flavipin | Polyketide | Aspergillus flavipes, Aspergillus terreus, Aspergillus fumigatus | |
| (3S,6S)-Terramide A | Diketopiperazine alkaloid | Aspergillus terreus, Aspergillus flavus | |
| 3-Methylorsellinic acid | Phenolic acid | Aspergillus terreus | |
| Terremutin hydrate | Dihydrotoluquinones | Aspergillus terreus | |
| Poh 3 | Class I hydrophobins | Pleurotus ostreatus | [112] |
| Epi-phelligrin A | Phenylpropanoids and polyketides | Sanghuangporus baumii | |
| Sterenin M | Isoprenylated depside | Stereum hirsutum | |
| Termitomycamide B | Indoles | Termitomyces titanicus | |
| Enokipodin D | Quinones | Flammulina velutipes | |
| Chondrillasterol | Steroid | Paenibacillus dendridiformis | [113] |
| Cholestan | Sterol lipids | Paenibacillus dendridiformis | |
| Trifluoroacetic acid | Organofluorine | Paenibacillus dendridiformis | |
| Octadeccenoic-acid | Glycerol ester | Bacillus subtilis | |
| Stigmasterol | Stigmastanes | Paenibacillus dendridiformis | |
| Octadecenoic acid | Monocarboxylic acid | Paenibacillus dendridiformis | |
| Hexadecanoic acid | Fatty acid | Bacillus subtilis | |
| Apratoxin A | Cyclodepsipeptide | Cyanobacterial lyngbya spp. | [114] |
| Carrageenan | Sulfated polysaccharides | Chondrus crispus | |
| Cryptophycin 52 | Dioxadiazacyclohexadecenetetrone cytotoxins | Nostoc cyanobacteria | |
| Cylindrospermopsin | Cyclic guanidine alkaloid | Cylindrospermopsis, Aphanizomenon, Anabaena, Lyngbya, Umezakia, and Raphidiopsis | |
| Deoxycylindrospermopsin | Triazaacenaphthylene | Cylindrospermopsis raciborskii | |
| Eucapsitrione | Anthraquinone | Cyanobacterium eucapsis sp. | |
| Tjipanazole A1 | Alkaloids | Tolypothrix tjipanasensis | |
| Tolyporphin | Tetrapyrroles | Tolypothrix nodosa | |
| Bacteriocin glycocin F | Peptide | Lactobacillus plantarum | [115] |
| Cathelidicin-6 | Peptide | Bos taurus | |
| Subtilosin-A | Peptide | Bacillus subtilis | |
| Bacteriocin PlnK | Peptide | Lactobacillus plantarum | |
| Moronecidin | Peptide | Morone saxatilis | |
| Bacteriocin lactococcin-G subunit beta | Peptide | Lactococcus lactis subsp. Lactis | |
| Crotamine Ile-19 | Peptide | Crotalus durissus ruruima | |
| Bacteriocin leucocin-A | Peptide | Leuconostoc gelidum | |
| M-zodatoxin-Lt2a | Peptide | Lachesana tarabaevi | |
| Polyphemusin-1 | Peptide | Limulus polyphemus | |
| Corticostatin-related peptide RK-1 | Peptide | Oryctolagus cuniculus | |
| Potential Inhibitor for PLpro-CoV2 | |||
| Fonsecin | Naphtho-gamma-pyrone | Aspergillus fonsecaeus | [116] |
| Pyranonigrin-B | Pyranopyrroles | Aspergillus niger LL-LV3020 | |
| Nigerloxin | Benzoic acid | Aspergillus niger | |
| Flaviolin | Naphthoquinones | Aspergillus sp. | |
| Tensidol A | Furopyrrols | Aspergillus niger | |
| Ochratoxin Beta | Fungal metabolites | Aspergillus ochraceus | |
| Altertoxin V | Perylene quinone | Alternaria tenuissima | [110] |
| Altertoxin II | Perylene quinone | Alternaria tenuissima | |
| Penicillixanthone A | Xanthone dimer | Aspergillus fumigates | |
| Cytochalasin Z8 | Cytochalasan alkaloid | Spicaria elegans | |
| Stachybotrosin D | Alcoholic O-sulfation | Stachybotrys chartarum | |
| Chloropupukeanolide A | Spiroketal peroxides | Pestalotiopsis fici | |
| Phomasetin | Alkaloids | Phoma sp. | |
| Isochaetochromin D1 | Polyketide | Fusarium sp. | |
| Aspergilol H (R) | Anthraquinones | Aspergillus versicolor | |
| Aspergilol H (S) | Anthraquinones | Aspergillus versicolor | |
| 11a-Dehydroxyiso-terreulactone A | Terpenoid | Aspergillus terreus SCSGAF0162 | |
| Arisugacin A | Aromatic ether organic heterotetracyclic | Penicillium sp. | |
| Aspernolide A | Butyrolactone secondary metabolite | Cladosporium cladosporioides | |
| Rhodatin | Meroterpenoid | Rhodotus palmatus | |
| Scedapin C | Fumiquinazoline alkaloids | Scedosporium apiospermum | |
| Scequinadoline A | Fumiquinozalines | Dichotomomyces cejpii | |
| 14S-Oxoglyantrypine | Indole alkaloid | Cladosporium sp. PJX-41 | |
| Deoxynortryptoquivaline | Quinazoline alkaloid | Aspergillus clavatus | |
| Quinadoline B | Alkaloid | Cladosporium sp. PJX-41 | |
| Norquinadoline A | Fumiquinazoline alkaloids | Cladosporium sp. PJX-41 | |
| Asperterrestide A (S) | Cyclic tetrapeptide | Aspergillus terreus SCSGAF0162 | |
| Asperterrestide A (R) | Cyclic tetrapeptide | Aspergillus terreus SCSGAF0162 | |
| Rubrolide S | Rubrolide | Aspergillus terreus OUCMDZ-1925 | |
| Isoaspulvinone | Aspulvinone | Aspergillus terreus | |
| Fulvic acid | Organic acid | Many microorganisms | |
| Cryptophycin 1 | Peptolides | Nostoc sp. ATCC 53789, Nostoc sp. GSV 224. | [114] |
| Cryptophycin 52 | Peptolides | Nostoc sp. | |
| Deoxycylindrospermopsin | Triazaacenaphthylene | Cylindrospermopsis raciborskii | |
| Fijimycin A | Cyclic depsipeptide | Streptomyces sp. | [117] |
| Kocurin | Thiazolyl peptide | Kocuria palustris | |
| Cyclosporin A | Cyclic non-ribosomal peptides | Beauveria nivea | |
| Dactinomycin | Chromopeptide antineoplastic antibiotic | Streptomyces parvulus | |
| Daptomycin | Lipopeptide antibiotic | Streptomyces roseosporus | |
| Emericellamides A | Cyclodepsipeptide | Emericella sp. | |
| Trichoderin | Mycobacterial aminolipopeptide | Trichoderma sp. | |
| Marthiapeptide | Polythiazole cyclopeptide | Brevibacillus sp. | |
| Leodoglucomide | Microbial non-ribosomal peptide | Bacillus licheniformis | |
| Unguisin | Cyclic heptapeptide | Emericella unguis | |
| Lajolamycin | Microbial non-ribosomal peptide | Streptomyces nodosus | |
| Brunsvicamide A | Cyclic peptide | Tychonema sp. | |
| Tyrocidine A | Cyclic decapeptide | Bacillus brevis | |
| 11-O-methylpseurotin A | Fungal metabolite | Aspergillus fumigatus | |
| Lobocyclamide B | Cyclododecapeptide | Lyngbya confervoides | |
| Ngercheumicin I | Cyclic depsipeptide | Photobacterium sp. | |
| Nocathiacins I | Cyclic thiazolyl peptides | Nocardia sp. | |
| Solonamide A | Non-ribosomal depsipeptide | Photobacterium sp. | |
| Thiocoraline | Cyclic depsipeptide | Micromonospora. I. | |
| Gramicidin S | Cyclic decapeptide | Bacillus brevis | |
| Structure | Compound/Peptide | Method Analysis | Binding Energy (kcal/mol) | Ref. |
|---|---|---|---|---|
| Potential Inhibitor for 3CLpro-CoV2 | ||||
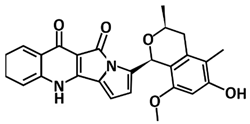 | Citriquinochroman | Pharmacophore-based virtual screening using Pharmit molecular docking Autodock Vina | −14.7 | [108] |
 | Pyranonigrin A | Molecular docking Autodock Vina, molecular dynamics using GROMACS 2019, ADMET analysis pkCSM–pharmacokinetics server | −7.3 | [109] |
 | 11a-dehydroxyisoterreulactone A | Molecular docking using UCSF Chimera, molecular dynamics using Amber 18, computational prediction of the absorption, distribution, metabolism, and excretion (ADME) properties using SwissADME software | −8.9 | [110] |
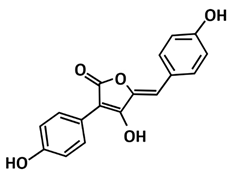 | Aspergillide B1 | Molecular docking using OpenEye’s FRED | −9.473 | [111] |
 | 3α-Hydroxy-3,5-dihydromonacolin L | Molecular docking using OpenEye’s FRED | −9.386 | [111] |
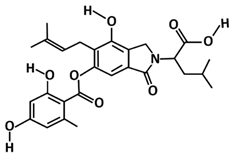 | Sterenin M | Molecular docking using Glide, ADMET analysis pkCSM–pharmacokinetics server, molecular dynamics using Desmond | −8.431 | [112] |
 | Hexadecanoic acid | Molecular docking using AutoDock, molecular dynamics using Amber 18 | −6.9 | [113] |
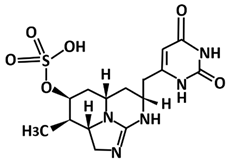 | Deoxycylindrospermopsin | Molecular docking using AutoDock Vina, molecular dynamics using GROMACS 2019 | −8.6 | [114] |
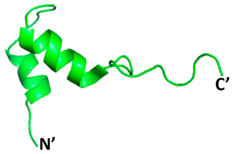 PDB ID: 2KUY | Bacteriocin glycocin F | Molecular docking using UCSF Chimera, molecular dynamics using GROMACS | −155.3 | [115] |
| Potential Inhibitor for PLpro-CoV2 | ||||
 | Fonsecin | Molecular docking using AutoDock Vina, ADMET analysis pkCSM–pharmacokinetics server | −7.25 | [116] |
 | Scedapin C | Molecular docking using UCSF Chimera, molecular dynamics using Amber 18, computational prediction of the absorption, distribution, metabolism, and excretion (ADME) properties using SwissADME software | −10.9 | [110] |
 | Norquinadoline A | Molecular docking using UCSF chimera, molecular dynamics using Amber 18, computational prediction of the absorption, distribution, metabolism, and excretion (ADME) properties using SwissADME software | −10.9 | [110] |
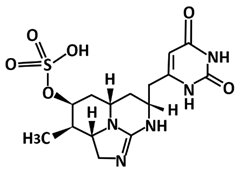 | Deoxycylindrospermopsin | Molecular docking using AutoDock Vina, molecular dynamics using GROMACS 2019 | −7.9 | [114] |
 PDB ID: 4M6E | Tyrocidine A | Molecular docking using AutoDock Vina | −13.1 | [117] |
 PDB ID: 1TK2 | Gramicidin S | Molecular docking using AutoDock Vina | −11.4 | [117] |
| Compound | Type | Binding Energy (kcal/mol) | Ref. |
|---|---|---|---|
| Plant Compounds Screened Against 3CLpro-CoV2 | |||
| 10-hydroxyusambarensine | Indole alkaloid | −10.0 | [118] |
| Cryptoquindoline | Cryptolepine | −9.7 | |
| Cryptospirolepine | Cryptolepine | −9.1 | |
| Chrysopentamine | Indole alkaloid | −8.6 | |
| Isocryptolepine | Cryptolepine | −8.5 | |
| Strychnopentamine | Indole akaloid | −8.2 | |
| Isostrychnopentamine | Indole akaloid | −8.1 | |
| Normelicopicine | Acridone | −8.1 | |
| Jozipeltine A | Naphthoisoquinoline | −8.0 | |
| 5′-O-demethyl-dioncophylline A | Naphthoisoquinoline | −8.0 | |
| Dioncophylline C | Naphthoisoquinoline | −7.9 | |
| Dioncopeltine A | Naphthoisoquinolines | −7.8 | |
| Liriodenine | Indole alkaloid | −7.6 | |
| 5,6-dihydronitidine | Furoquinoline | −7.6 | |
| Hydroxycryptolepine | Cryptolepine | −7.6 | |
| Cryptoheptine | Cryptolepine | −7.6 | |
| Annonidine F | Indole alkaloid | −7.5 | |
| Ancistrotanzanine C | Naphthoisoquinoline | −7.5 | |
| Fagaronine | Indole alkaloid | −7.4 | |
| Alstonine | Indole alkaloid | −7.4 | |
| Curcumin | Curcuminoid | −6.04 | [119] |
| Bisdemethoxycurcumin | Curcuminoid | −7.3 | |
| Demethoxycurcumin | Curcuminoid | −7.02 | |
| Scutellarin | Flavanoid | −7.13 | |
| Quercetin | Flavanoid | −6.58 | |
| Myricetin | Flavanoid | −6.15 | |
| Bergapten | 5-methoxypsoralen | 5.98 | |
| Isoflavone | Flavanoid | 5.69 | |
| Spicatolignan | Lignan | −6.7403 | [120] |
| Vanillic acid | Benzenoid | −4.8624 | |
| Ferulic acid | Hydroxycinnamic acid | −5.3292 | |
| Pinoresinol | Furanoid ligan | −6.463 | |
| Sesamolin | Lignan | −6.829 | |
| Sesamin | Lignan | −6.7157 | |
| Hydroxymatairesinol | Lignan | −7.4674 | |
| Saikosaponin D | Saikosaponin | −8.9 | [121] |
| Saikosaponin E | Saikosaponin | −8.9 | |
| Myricetin | Flavanoid | −8.9 | |
| Theaflavin | Catechin | −8.6 | |
| Glycyrrhizin | Triterpenoid saponin | −8.7 | |
| Plant compounds screened against PLpro-CoV2 | |||
| Hydroxymatairesinol | Lignan | −7.2085 | [120] |
| Spicatolignan | Lignan | −6.6183 | |
| Vanillic acid | Benzenoid | −4.6805 | |
| Ferulic acid | Hydroxycinnamic acid | −4.8177 | |
| Pinoresinol | Furanoid ligan | −6.5131 | |
| Sesamolin | Lignan | −6.454 | |
| Sesamin | Lignan | −6.5524 | |
| Amentoflavone | Biflavonoid | −9.2 | [121] |
| Glycyrrhizin | Triterpenoid saponin | −9.6 | |
| Theaflavin | Biflavonoid | −9.1 | |
| Chrysin-7-O-glucuronide | Flavonoid | −8.8 | |
| Isoquercitrin | Flavonoid-3-O-glycoside | −8.5 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razali, R.; Asis, H.; Budiman, C. Structure-Function Characteristics of SARS-CoV-2 Proteases and Their Potential Inhibitors from Microbial Sources. Microorganisms 2021, 9, 2481. https://doi.org/10.3390/microorganisms9122481
Razali R, Asis H, Budiman C. Structure-Function Characteristics of SARS-CoV-2 Proteases and Their Potential Inhibitors from Microbial Sources. Microorganisms. 2021; 9(12):2481. https://doi.org/10.3390/microorganisms9122481
Chicago/Turabian StyleRazali, Rafida, Haslina Asis, and Cahyo Budiman. 2021. "Structure-Function Characteristics of SARS-CoV-2 Proteases and Their Potential Inhibitors from Microbial Sources" Microorganisms 9, no. 12: 2481. https://doi.org/10.3390/microorganisms9122481
APA StyleRazali, R., Asis, H., & Budiman, C. (2021). Structure-Function Characteristics of SARS-CoV-2 Proteases and Their Potential Inhibitors from Microbial Sources. Microorganisms, 9(12), 2481. https://doi.org/10.3390/microorganisms9122481