Abstract
Helicobacter pylori is well established as a causative agent for gastritis, peptic ulcer, and gastric cancer. Armed with various inimitable virulence factors, this Gram-negative bacterium is one of few microorganisms that is capable of circumventing the harsh environment of the stomach. The unique spiral structure, flagella, and outer membrane proteins accelerate H. pylori movement within the viscous gastric mucosal layers while facilitating its attachment to the epithelial cells. Furthermore, secretion of urease from H. pylori eases the acidic pH within the stomach, thus creating a niche for bacteria survival and replication. Upon gaining a foothold in the gastric epithelial lining, bacterial protein CagA is injected into host cells through a type IV secretion system (T4SS), which together with VacA, damage the gastric epithelial cells. H. pylori does not only establishes colonization in the stomach, but also manipulates the host immune system to permit long-term persistence. Prolonged H. pylori infection causes chronic inflammation that precedes gastric cancer. The current review provides a brief outlook on H. pylori survival tactics, bacterial-host interaction and their importance in therapeutic intervention as well as vaccine development.
1. Introduction
Helicobacter pylori is a Gram-negative bacterium that selectively colonizes the gastric epithelium. It is classified in the order of Campylobacterales and the family of Helicobacteraceae. The term “Helico-” is of Greek origin and means spiral or curved, representing the unique shape of H. pylori. In 1994, H. pylori was recognized as a Type I carcinogen by the World Health Organization (WHO) [1,2]. Infection commonly results in chronic gastritis that eventually leads to intestinal metaplasia and dysplasia, which in turn culminate in the initiation of gastric cancer [3,4]. It has been reported that more than 90% of the gastric cancer patients are infected with H. pylori [5], and that 6.2% of all human cancer cases occurring worldwide are attributable to its infection [6]. Approximately 15–20% of the infected individuals develop severe gastroduodenal pathologies, among whom 1–2% progress to gastric cancer during their lifetime [7]. A recent systemic review also suggested a pooled prevalence of up to 17.4% of Helicobacter pylori-mediated gastric cancers among infected population [8].
It is estimated that 50% of the world’s population is infected with H. pylori. The infection is usually acquired during childhood and can persist to adulthood when left untreated [9]. In a systematic review conducted by Hooi, Lai [10], the highest prevalence of H. pylori was reported in Africa (70.1%), followed by South America (69.4%) and Western Asia (66.6%), whereas the lowest prevalence was observed in Oceania (24.4%), Western Europe (34.3%), and Northern America (37.1%). These data denote lower socioeconomic status and poor hygiene practices as the risk factors for H. pylori infection [11]. Currently, the histological staining of stomach biopsy is a gold standard for diagnosis. The first-line treatment remains triple therapy using clarithromycin, amoxicillin or metronidazole, and proton pump inhibitor, although increasing antibiotic resistance of bacteria necessitates further improvement of the current therapeutic strategy [12,13].
H. pylori is one of the most successful human pathogens and has coexisted and adapted to humans for at least 50,000 years. This review discusses the unique bacterial characteristics that confer its successful survival within the stomach lumen. The unique helical shape, ability to produce urease, flagella movement, and outer membrane proteins (OMPs) are among the contributing factors [14]. Upon reaching the gastric epithelium, H. pylori attaches firmly to host cells via various OMPs and produces a permanent colonizing strain to prevent detachment from the peristaltic movement of the bowels. Then, it induces gastric mucosal damage via its virulence proteins, CagA and VacA. Damage to the gastric epithelial cells following bacteria colonization induces chronic inflammatory reaction, leading to gastritis, peptic ulcer, or gastric carcinoma. The following sections discuss the key features of H. pylori that determine a successful colonization of the gastric mucosa (Figure 1).
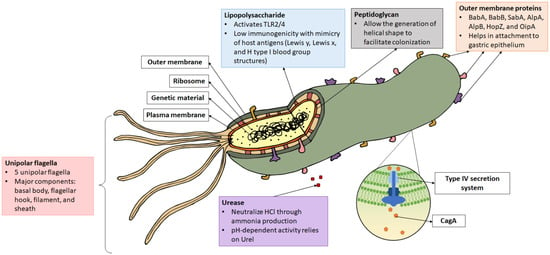
Figure 1.
Structure of Helicobacter pylori. Virulence factors such as lipopolysaccharide (LPS), flagella, urease, peptidoglycan, outer membrane protein, CagA, and the type IV secretion system (T4SS) are as indicated. CagA is injected into the host cell via T4SS.
1.1. H. pylori Structures Facilitate Bacterial Motility in the Thick Mucosal Layers
H. pylori is a good swimmer that can travel through the viscous mucus layer like a screw driven into a cork. It is approximately 0.5 µm in diameter and 3–5 µm long, with one to three spiral turns. Its helical shape, formed by peptidoglycan structural arrangement and cross-linking, enables the bacterium to burrow deeper into the mucus layer [15]. It has been shown that the first colonizing strain of H. pylori establishes an irreplaceable “founder colony”, which is found deep in the gastric glands, serving as a reservoir to the transient population in the superficial mucosa that is shed due to peristalsis [16]. In addition, H. pylori is armed with four to six unipolar flagella. Each flagellum measures around 30 nm in diameter and 12–15 nm in length. One flagellum is made up of several subunits: flagellar basal body, flagellar hook, flagellar filament, and flagellar sheath [17]. The expression, synthesis, and finally assembly of a single flagellum has been estimated to involve more than 50 different putative proteins. Mutations in genes that form the filament (flaA encoding the major structural component and flaB expressing the minor species that localizes to the base of the flagellum) and flagellar hook (flgE encoding the structural protein and fliD for hook-associated protein expression) have been reported to cause the formation of truncated flagella or result in aflagellated strains with reduced colonizing abilities [18]. Moreover, recent evidence has shown the importance of flagella in the formation of bacterial biofilm to enhance persistence. Among the involved genes are flgB (encoding rod protein), flgE, flgK and flgL (both encoding hook-filament junction proteins), fliK (encoding hook length control protein), and flaB and flag (encoding filament protein) [19].
1.2. OMPs Facilitate Bacterial Attachment to the Gastric Epithelial Cells
The outer membrane proteins (OMPs) provide a barrier for the bacterium to resist its external environment. Furthermore, OMPs have been shown to be an important component for bacteria attachment to host cells. A total of 4% of the bacterial genome encodes for a diverse family of OMPs in H. pylori, which is twice the amount compared to E. coli [20,21]. The largest member of the OMPs is the Helicobacter outer membrane porin (Hop) family. Among the well-studied Hop proteins are HopS and HopT (also named as blood antigen-binding proteins, BabA and BabB), which facilitate the attachment of bacteria to host cells via binding with the Lewis b (Leb)-histo-blood group antigen [22]. HopP (also called sialic acid-binding adhesin, SabA) binds to the inflammation-associated sialyl-dimeric-Lewis ßods x glycosphingolipid on gastric epithelial cells during chronic inflammation, leading to enhanced colonization [23]. HopC (AlpA) and HopB (AlpB) bind to laminin in the host cell; their absence in H. pylori SS1 mutant caused a more severe inflammation in gerbils [24]. HopC, HopB, HopZ, and HopH (also named as outer inflammatory protein A, OipA) are also among the OMPs that are indispensable during bacterial attachment and colonization in the stomach of experimental guinea pig [25]. It has been reported that the H. pylori OMP HopH/OipA can suppress dendritic cell maturation and interleukin-10 (IL-10) secretion, dampening immune activation [26]. Apart from this, OipA can induce apoptosis in gastric epithelial cells via Bax/Bcl-2 modulation [27]. Another molecule identified to be involved in immune modulation is HopQ, which interacts with CEA Cell Adhesion Molecule 1 (CEACAM1)-expressing CD4+ T cells to reduce interferon gamma (IFN-γ) production; CEACAM1-expressing CD8+ T cells, and CD16– natural killer cells to inhibit cytotoxicity [28].
1.3. H. pylori Urease Neutralizes Acidic pH
Several genes in the genome of H. pylori are dedicated to the production of urease, which hydrolyses urea into ammonia (NH4) and carbon dioxide (CO2). Urease is typically found on the bacterial surface or in the cytoplasm that is released upon bacteria lysis. A urease-negative H. pylori strain showed impaired colonizing ability and was unable to be isolated from gnotobiotic piglets following 3 days of infection [29]. Additionally, expression of urease was shown to be important for persistence of bacteria using a conditional urease knockout strain of H. pylori [30]. Urea transporter (Urel) transports urea in a pH-dependent manner for ammonia production to buffer the bacterium’s periplasm, forming a neutral layer favorable to its survival. Not only does the NH4 produced function as a buffer, it is also known to be a toxic substance that causes damage to the host cells [18,31]. Interestingly, the urease-producing capacity of H. pylori also limits its ecological niche to the stomach, where optimal proton motive force can only be maintained in pH 3.5 to 8.4 in vitro to drive adenosine triphosphate (ATP) generation essential for its survival [32]. Besides urease’s ability to directly inflict damages, it is also a chemotactic agent that can recruit immune cells [33]. Furthermore, it has been shown to be capable of inducing angiogenesis using an AGS cell model, implying its contribution to gastric cancer development [34].
1.4. H. pylori Evades Host Immune Response
The ability of H. pylori to modulate host immunity is acquired through its long course of coevolution with humans. Among these survival tactics is the synthesis of bacterial components of low immunogenicity. For example, lipopolysaccharide (LPS) and flagellin derived from H. pylori have been reported to be weakly immunogenic [35,36]. Activation of TLR4 and TLR5 are reduced as a result, decreasing the activation of immune cells and inflammation via nuclear factor-kappa B (NF-ĸB) nuclear translocation [37,38]. Consequently, the host is unable to mount a strong immune response to rapidly eliminate the bacteria.
Furthermore, H. pylori exploits Mincle and Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Nonintegrin (DC-SIGN) in macrophages and dendritic cells to induce anti-inflammatory IL-10 secretion, and suppress both proinflammatory IL-12 and IL-6 production [39]. Additionally, H. pylori has been found to attenuate macrophage proliferation in an in vitro setting [40], resist phagocytosis and reactive oxygen species-mediated bacterial killing [41,42], thereby enabling persistent infection. It is also able to hinder expression of human leukocyte antigen-II (HLA-II) and IFN-γ production from macrophages, which in turn decreases T cell activation [43]. Apart from this, infection with H. pylori induces the recruitment of dendritic cells that produce transforming growth factor-β (TGF-β) and IL-10. This skews the T cells’ response towards a more regulatory phenotype [44]. It was further observed that expression of coinhibitory molecules including program death-ligand 1 (PD-L1) was elevated in both dendritic cells and CD4+ T cells, leading to T cell anergy and impaired bacterial clearance [45,46]. More recently, it is shown that H. pylori and IL-22 induce metalloprotease-10 (MMP-10) expression in gastric epithelial cells, which enhances bacterial colonization and associated pathology via reduction in antibacterial protein Reg3a production, decrement of tight junction proteins including E-cadherin and ZO-1, and enhancement of inflammation indicated by the influx of CD8+ T cells [47].
1.5. Type IV Secretion System Penetrates Gastric Epithelial Cells
The type IV secretory system (T4SS) is a large transporter complex expressed on the surface of Gram-negative bacteria and archaea to facilitate the transportation of proteins and DNA into host cell in a contact-dependent manner [48]. In H. pylori, it is encoded by the 40 kb genetic locus of cytotoxin-associated gene pathogenicity island (cagPAI), a portion of the chromosome that possesses different CG content and is usually acquired through horizontal transfer. It has a 41 nm-long core structure consisting of different bacterial proteins including CagM, CagT, Cag3, CagX, and CagY, which protrude from the bacterial surface into the host cell. CagX and CagY are orthologous bacterial components that form the putative channel of the T4SS [49]. CagL is important for anchoring the T4SS to the adhesion molecule α5β1 integrin on the epithelial cell [50]. It also binds to fibronectin, which induces cell spreading, focal adhesion formation, and activation of cell tyrosine kinases that facilitate CagA pathogenesis [51]. Additionally, T4SS is known to transport HopS/BabA to increase the production of proinflammatory and precancer-related factors [52]. A functional T4SS has also been recognized to induce IL-18 production in the gastric epithelial cells via NLRC4 inflammasome activation, causing bacterial persistence and enhanced inflammation in mice [53].
1.6. CagA Perturbs Normal Cell Activities
CagA is a hydrophilic, surface-exposed protein that may be present or absent in H. pylori. Due to its importance in the outcome of infection, classification based on the presence of the CagA-encoding cagPAI results in the identification of cytotoxin-associated gene A (CagA) positive, Cag+, or negative, Cag− H. pylori strains [54]. In Egypt, the presence of Cag+ H. pylori was detected among 33.3% of patients with gastritis, 68.7% of peptic ulcer cases, and 50% of gastric carcinoma cases [55]. Meanwhile, the Austrian population that revealed low H. pylori prevalence among patients with duodenal ulcers (20.8%) and gastric cancer (16.6%) found Cag+ strains among 78% and 85% of the isolated H. pylori strains from respective diseases [56]. With high variability among different Cag+ H. pylori, two different variants of CagA have been described based on binding affinity to SHP2, namely, the East Asian CagA, with stronger binding affinity and higher pathogenicity, and the Western CagA, with lower binding affinity that makes it less virulent than the former [57].
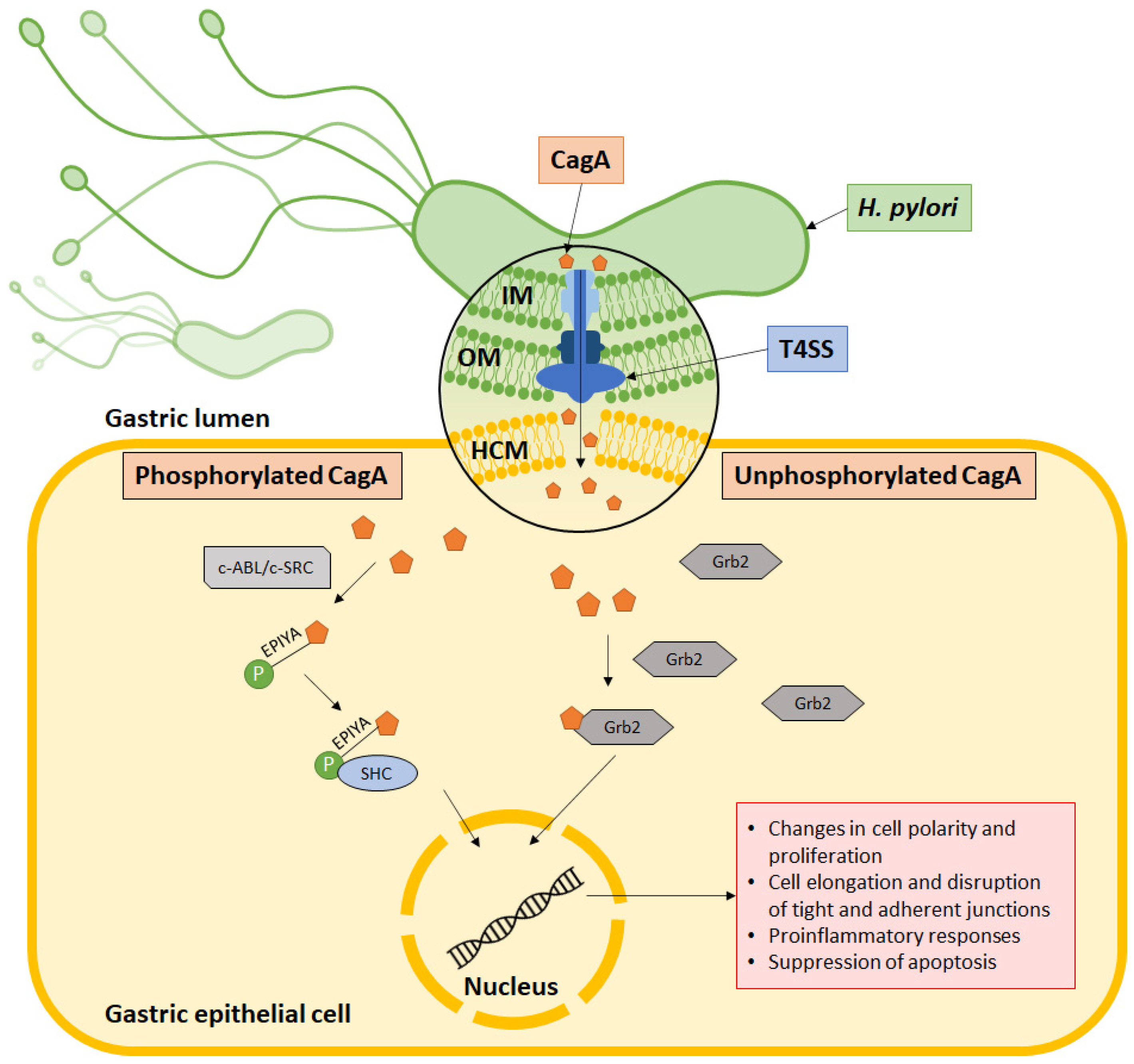
Within the cell, CagA is phosphorylated at the C-terminal EPIYA motif by host c-SRC and c-ABL kinases. It then binds to the SH2 domain to activate a series of oncogenic signaling processes (Figure 2). This activates signaling pathways that subsequently cause aberrant changes to cell polarity, cell proliferation, actin-cytoskeletal rearrangements, cell elongation, disruption of tight and adherent junctions, proinflammatory responses, and suppression of apoptosis. On the other hand, nonphosphorylated CagA interacts with several other cellular components, such as Grb2, which leads to loss of cell polarity, mitogenic responses, and proinflammatory signaling [58,59]. In its molecular involvement during carcinogenesis, CagA protein was identified to inhibit PAR1b-mediated BRCA1 phosphorylation, enhance DNA double breaks, and stimulate Hippo signaling, all of which drive genome instability during development of cancer-predisposing cells [60]. Activation of YAP signaling by CagA was further observed to promote epithelial mesenchymal transition in the gastric epithelial cells, thereby accelerating carcinogenesis and cancer dissemination [61].
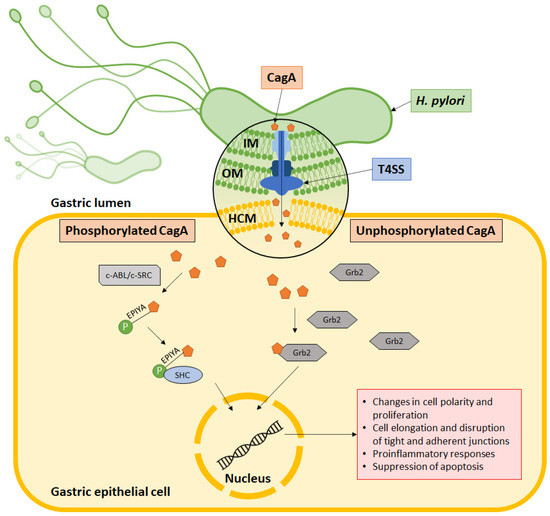
Figure 2.
CagA’s mechanism of action. CagA is injected into the host cell via the type IV secretion system (T4SS), which is made up of an outer membrane ring occupying the outer membrane (OM), an inner membrane ring at the inner membrane (IM) and a pilus to inject CagA across the host cell membrane (HCM) into the cytoplasm. Some CagA undergo phosphorylation at the EPIYA motif via c-ABL or c-SRC and associates with the SHC domain to induce pathologies. Meanwhile, some CagA associate with Grb2 to induce pathology without undergoing phosphorylation.
1.7. VacA Induces Host Cell Vacuolation
VacA is a toxin secreted by H. pylori to induce the formation of large cytoplasmic vacuoles in host cells. It has two functional domains that are linked together via a hydrophilic loop, namely the p58 domain, which allows host cell binding, and the N-terminal p33 domain, which is responsible for vacuole formation [62,63]. Similar to cagA, the vacA gene is highly polymorphic across different H. pylori strains and contributes to variable disease outcomes. There are two different families within each vacA sequence: s1 and s2 within signal sequences, i1 and i2 in intermediate regions, and m1 and m2 in middle-region sequences. Type s1/m1 strain produces higher levels of cytotoxic and inflammatory activity in the gastroduodenal region [64,65].
Structurally, VacA is heat-labile, protease-sensitive and assume an oligomeric structure that dissociates upon acid or alkaline exposure. This process activates the toxin and potentiates its binding to host cell surface receptor-type protein tyrosine phosphatase β [66]. The toxin remains highly activated at pH 1.5 to 6 and is strongly resistant against pepsin digestion [67]. VacA can bind to the bacterial surface as an active monomeric form that is delivered into host cells upon bacterial adhesion [68]. When inserted into the apical plasma membrane of the gastric epithelial cells, it alters cell-cell interactions and increases in the permeability of nutrients essential for the growth of H. pylori from the underlying mucosa [69].
During carcinogenesis, VacA causes a decrease in trans-epithelial resistance and a concomitant increase in paracellular permeability [70,71,72]. It also effectively impairs mitochondria function and inhibits mammalian target of rapamycin complex 1 (mTORC1) to dysregulate the cellular metabolism of gastric epithelial cells. This leads to cellular autophagy that potentially impedes immune effector production [73]. VacA also enhances TGF-β1 production, which subsequently induces inflammatory response during gastritis [74]. It has also been shown to be able to target the endoplasmic reticulum stress pathway to induce AGS autophagy [75].
VacA-induced vacuolization causes a marked decrease in antigen proteolysis among antigen-presenting cells, a process that is required to generate peptide epitopes. The decrease in proteolytic activity reduces peptide presentation, consequently inhibiting the stimulation of T cells [69]. There has been evidence that persistent H. pylori infection decreases specific CD8+ cytotoxic T cell and CD4+ helper T cell activities, leading to depressed local immune response [76,77]. In addition, VacA inhibits T cell proliferation at the G1/S phase through abrogation of nuclear factor of activated T cells (NFAT) translocation, thus effectively suppressing the production of T cell growth factor IL-2 [78].
1.8. H. pylori as a Causative Agent of Gastric Cancer
H. pylori infection causes a typical series of events that eventually leads to gastric cancer in a progression cascade proposed by Correa, Haenszel [79]. The cascade starts from an inflammatory response of the gastric mucosa in the presence of H. pylori infection. This is followed shortly by acute gastritis that slowly becomes chronic gastritis. Chronic gastritis worsens, which leads to atrophic gastritis. The loss of gastric glandular cells causes increased stomach pH. Altered conditions in the stomach together with other genetic and environmental factors contribute to the development of intestinal metaplasia, then dysplasia, and finally gastric cancer. Because of the lack of specific symptoms during the early stages of gastric cancer, patients are usually diagnosed only after the cancer has invaded the muscularis propria. This may be one of the contributing factors as to why the 5-year survival rate for gastric cancer is less than 15% in the United States [80].
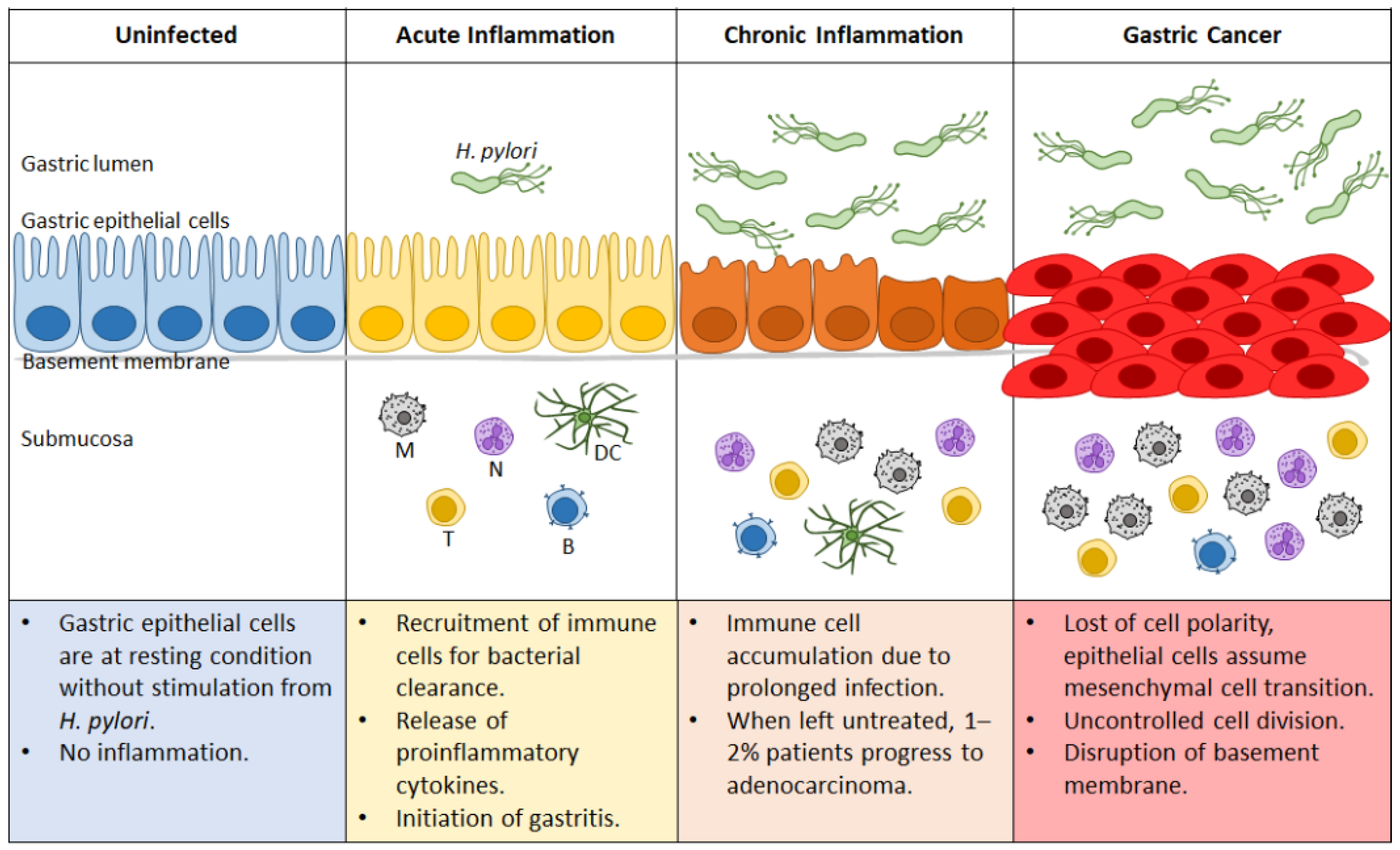
At early timepoints, most infected patients are unaware of the bacterium’s colonization because of its asymptomatic manifestations. The initial response to the infection is marked by an increase in highly inflammatory immune cells, which are unfortunately ineffective at H. pylori elimination because of the bacterium’s various immune evasion strategies. Hence, the recruitment of these cells results in epithelial damage instead of the expected eradication. The persistent presence of H. pylori thus leads to both the chronic proinflammatory response and cellular damage that contribute to the development of cancer. Figure 3 summarizes the role of H. pylori in the transformation of an uninfected healthy gastric epithelium to the state of gastric cancer.
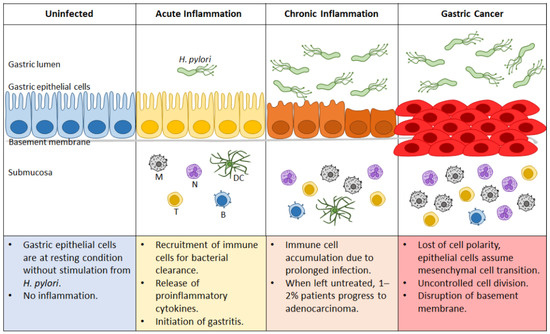
Figure 3.
Progression of gastric cancer during Helicobacter pylori infection. Uninfected gastric epithelial cells are healthy, with no sign of inflammation. Upon H. pylori infection, acute inflammation ensues that can progress to gastritis. Ineffective bacterial clearance leads to prolonged infection and continuous recruitment of immune cells, resulting in chronic inflammation. Depending on genetic and environmental factors, approximately 1–2% of the afflicted patients progress to gastric cancer. N: neutrophil; M: macrophage; DC: dendritic cell; T: T cell; B: B cell.
1.9. Targeting H. pylori Virulence and Pathogenesis in Treatment and Vaccine Development
The discovery of Helicobacter pylori as a causative agent for gastritis and gastric cancer has geared the direction of therapeutic strategy towards antimicrobial regimens. However, attempts to achieve a complete elimination of the bacteria using the devised proton-pump inhibitors-based triple therapy has proven to be challenging, partly because of increasing antibiotic resistance [12]. Furthermore, a growing knowledge of the various H. pylori virulence factors during its pathogenesis has not significantly altered the management of the disease from the decades-old antibiotic regimen. To date, gastric cancer is still the fifth most common and fourth most deadly cancer worldwide [81], with the highest number of cases contributed by China, Japan, and Korea [82,83,84]. Hence, further understanding of the survival tactics and pathogenesis of H. pylori is crucial for the rational development of a better curative method.
Various alternatives targeting H. pylori virulence and interaction with host cells have been explored, albeit with little translation into the clinical setting. Among these targets, drugs against H. pylori urease production have been popularly sought after, as it contributed significantly to bacterial survival advantage and immune activation [85]. Furthermore, T4SS has also long been proposed as a treatment target, as inhibitors may be able to block the injection of virulence proteins such as CagA into host cells [86]. In a recent report, an in silico genome-scale protein interaction network was constructed, potentiating the identification of therapeutic targets [87]. Targeting H. pylori-host interaction has also seen major advancement, especially in the alleviation of chronic inflammation. It has been identified that H. pylori induces expression of an inflammatory protein, podoplanin, via LPS to induce strong proinflammatory IL-1β secretion from macrophages [88]. Podoplanin has been proposed as a treatment target in chronic inflammatory rheumatoid arthritis [89]. Hence, it is not too far-fetched to explore this possibility in the treatment of H. pylori chronic infection. Other host molecules that have been identified include heparanase, an enzyme that has been exploited to facilitate immune cell recruitment and inflammation during infection [90]. Inhibition of hepatoma-derived growth factor has been proposed to reduce the degree of inflammation and immune cell recruitment [91]. Other than this, disrupting the pathways activated by H. pylori during the initiation of gastric cancer has also been suggested. These pathways include COX-2/Wnt/beta-catenin/VEGF, TLR2/TLR9/COX-2, COX2-PGE2, and NF-κB/COX-2, as well as EPHA2, MMPs, and the miR-543/SIRT1 axis [92].
An effective vaccine has been in active search with the hope of reducing severity and prevalence of H. pylori infection, especially in developing countries. Most of the existing vaccine candidates are made up of purified or recombinant components of H. pylori antigens with an adjuvant [93]. For instance, urease subunits (UreA and UreB) have gained significant attention as potential prophylactic and therapeutic vaccines. However, only one candidate has proceeded to Phase III clinical trial and demonstrated protection against natural acquisition of infection among children in a prospective study [94]. This aside, most of the vaccines targeting other virulence factors are either preclinical or in Phase I clinical trials. Among the targets are multiple adhesion molecules (SabA, BabA, H. pylori adhesion A subunit (HpaA)) and other bacterial factors (CagA, VacA neutrophil-activating protein (NAP), FlaA) [95].
2. Conclusions
In summary, understanding the virulence and pathogenesis of H. pylori continues to be relevant and important as our knowledge about this gut pathogen seems to continually expand. As discussed above, H. pylori can persist in the human stomach through establish of founder colonies, pH neutralization, gastric epithelial cell disruption, and host immune modulation. Exploration of the virulence targets may serve to accelerate vaccine research and provide better treatment alternatives to complement the existing antibiotic therapy.
Author Contributions
Conceptualization, W.F.W.; writing—original draft preparation, Y.Y.C., C.Y.Q.L. and H.C.C.; writing—review and editing, Y.Y.C. and W.F.W.; visualization, Y.Y.C.; supervision, C.Y.L., J.V. and S.A.; funding acquisition, S.A. and W.F.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Malaysia Ministry of Higher Education Fundamental Research Grant Scheme FRGS/1/2019/SKK06/UM/02/4 (FP133-2019A) and the Institut Mérieux Young Investigator Fund (IF039-2017). Y.Y. Cheok was funded by a Doctoral Scholarship from the Malaysia Public Service Department (JPA).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer; World Health Organization. Schistosomes, Liver Flukes and Helicobacter pylori: IARC Monographs on the Carcinogenic Risks to Humans; Vol 61 Lyon, France International Agency for Research on Cancer; World Health Organization: Geneva, Switzerland, 1994; pp. 177–240. [Google Scholar]
- Peleteiro, B.; Bastos, A.; Ferro, A.; Lunet, N. Prevalence of Helicobacter pylori infection worldwide: A systematic review of studies with national coverage. Dig. Dis. Sci. 2014, 59, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Díaz, P.; Valenzuela Valderrama, M.; Bravo, J.; Quest, A.F.G. Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front. Microbiol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Gong, E.J.; Chung, E.J.; Park, H.W.; Bae, S.E.; Kim, E.H.; Kim, J.; Do, Y.S.; Kim, T.H.; Chang, H.S.; et al. The Characteristics and Prognosis of Diffuse-Type Early Gastric Cancer Diagnosed during Health Check-Ups. Gut Liver 2017, 11, 807–812. [Google Scholar] [CrossRef]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- Conteduca, V.; Sansonno, D.; Lauletta, G.; Russi, S.; Ingravallo, G.; Dammacco, F. H. pylori infection and gastric cancer: State of the art (Review). Int. J. Oncol. 2013, 42, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Pormohammad, A.; Mohtavinejad, N.; Gholizadeh, P.; Dabiri, H.; Salimi Chirani, A.; Hashemi, A.; Nasiri, M.J. Global estimate of gastric cancer in Helicobacter pylori–infected population: A systematic review and meta-analysis. J. Cell. Physiol. 2019, 234, 1208–1218. [Google Scholar] [CrossRef]
- Koch, M.; Mollenkopf, H.-J.; Meyer, T.F. Macrophages recognize the Helicobacter pylori type IV secretion system in the absence of toll-like receptor signalling. Cell. Microbiol. 2016, 18, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Eusebi, L.H.; Zagari, R.M.; Bazzoli, F. Epidemiology of Helicobacter pylori infection. Helicobacter 2014, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Siddique, O.; Ovalle, A.; Siddique, A.S.; Moss, S.F. Helicobacter pylori Infection: An Update for the Internist in the Age of Increasing Global Antibiotic Resistance. Am. J. Med. 2018, 131, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Fischbach, L. Helicobacter pylori treatment in the era of increasing antibiotic resistance. Gut 2010, 59, 1143. [Google Scholar] [CrossRef] [PubMed]
- Abadi, A.T.B. Strategies used by helicobacter pylori to establish persistent infection. World J. Gastroenterol. 2017, 23, 2870–2882. [Google Scholar] [CrossRef] [PubMed]
- Sycuro, L.K.; Pincus, Z.; Gutierrez, K.D.; Biboy, J.; Stern, C.A.; Vollmer, W.; Salama, N.R. Peptidoglycan Crosslinking Relaxation Promotes Helicobacter pylori’s Helical Shape and Stomach Colonization. Cell 2010, 141, 822–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.; Tan, S.; Nakajima, M.; Skoog, E.C.; Camarillo-Guerrero, L.F.; Klein, J.A.; Lawley, T.D.; Solnick, J.V.; Fukami, T.; Amieva, M.R. High-resolution mapping reveals that microniches in the gastric glands control Helicobacter pylori colonization of the stomach. PLoS Biol. 2019, 17, e3000231. [Google Scholar] [CrossRef] [Green Version]
- Gu, H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr. Microbiol. 2017, 74, 863–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagoonee, S.; Pellicano, R. Helicobacter pylori: Molecular basis for colonization and survival in gastric environment and resistance to antibiotics. A short review. Infect. Dis. 2019, 51, 399–408. [Google Scholar] [CrossRef]
- Hathroubi, S.; Zerebinski, J.; Ottemann Karen, M.; Freitag Nancy, E.; Hengge, R.; Cover, T. Helicobacter pylori Biofilm Involves a Multigene Stress-Biased Response, Including a Structural Role for Flagella. MBio 2018, 9, e01973-18. [Google Scholar] [CrossRef] [Green Version]
- Bauwens, E.; Joosten, M.; Taganna, J.; Rossi, M.; Debraekeleer, A.; Tay, A.; Peters, F.; Backert, S.; Fox, J.; Ducatelle, R.; et al. In silico proteomic and phylogenetic analysis of the outer membrane protein repertoire of gastric Helicobacter species. Sci. Rep. 2018, 8, 15453. [Google Scholar] [CrossRef]
- Alm, R.A.; Bina, J.; Andrews, B.M.; Doig, P.; Hancock, R.E.W.; Trust, T.J. Comparative Genomics of Helicobacter pylori: Analysis of the Outer Membrane Protein Families. Infect. Immun. 2000, 68, 4155. [Google Scholar] [CrossRef] [Green Version]
- Oleastro, M.; Ménard, A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology 2013, 2, 1110–1134. [Google Scholar] [CrossRef] [Green Version]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Ångström, J.; Larsson, T.; Teneberg, S.; Karlsson, K.-A.; et al. Helicobacter pylori SabA Adhesin in Persistent Infection and Chronic Inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Senkovich, O.A.; Yin, J.; Ekshyyan, V.; Conant, C.; Traylor, J.; Adegboyega, P.; McGee, D.J.; Rhoads, R.E.; Slepenkov, S.; Testerman, T.L. Helicobacter pylori AlpA and AlpB Bind Host Laminin and Influence Gastric Inflammation in Gerbils. Infect. Immun. 2011, 79, 3106–3116. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, R.; Durrani, Z.; Rijpkema, S.G.; Kuipers, E.J.; van Vliet, A.H.M.; Kusters, J.G. Role of the Helicobacter pylori outer-membrane proteins AlpA and AlpB in colonization of the guinea pig stomach. J. Med Microbiol. 2004, 53, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Teymournejad, O.; Mobarez, A.M.; Hassan, Z.M.; Moazzeni, S.M.; Ahmadabad, H.N. In Vitro Suppression of Dendritic Cells by Helicobacter pylori OipA. Helicobacter 2014, 19, 136–143. [Google Scholar] [CrossRef]
- Teymournejad, O.; Mobarez, A.M.; Hassan, Z.M.; Talebi Bezmin abadi, A. Binding of the Helicobacter pylori OipA causes apoptosis of host cells via modulation of Bax/Bcl-2 levels. Sci. Rep. 2017, 7, 8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gur, C.; Maalouf, N.; Gerhard, M.; Singer, B.B.; Emgård, J.; Temper, V.; Neuman, T.; Mandelboim, O.; Bachrach, G. The Helicobacter pylori HopQ outermembrane protein inhibits immune cell activities. OncoImmunology 2019, 8, e1553487. [Google Scholar] [CrossRef]
- Eaton, K.A.; Brooks, C.L.; Morgan, D.R.; Krakowka, S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991, 59, 2470–2475. [Google Scholar] [CrossRef] [Green Version]
- Debowski, A.W.; Walton, S.M.; Chua, E.-G.; Tay, A.C.-Y.; Liao, T.; Lamichhane, B.; Himbeck, R.; Stubbs, K.A.; Marshall, B.J.; Fulurija, A.; et al. Helicobacter pylori gene silencing in vivo demonstrates urease is essential for chronic infection. PLoS Pathog. 2017, 13, e1006464. [Google Scholar] [CrossRef]
- Mégraud, F.; Neman-Simha, V.; Brügmann, D. Further evidence of the toxic effect of ammonia produced by Helicobacter pylori urease on human epithelial cells. Infect. Immun. 1992, 60, 1858–1863. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Rosberg, K.; Scott, D.R.; Rex, D.; Melchers, K.; Sachs, G. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology 1996, 111, 886–900. [Google Scholar] [CrossRef]
- Uberti, A.F.; Olivera-Severo, D.; Wassermann, G.E.; Scopel-Guerra, A.; Moraes, J.A.; Barcellos-de-Souza, P.; Barja-Fidalgo, C.; Carlini, C.R. Pro-inflammatory properties and neutrophil activation by Helicobacter pylori urease. Toxicon 2013, 69, 240–249. [Google Scholar] [CrossRef]
- Olivera-Severo, D.; Uberti, A.F.; Marques, M.S.; Pinto, M.T.; Gomez-Lazaro, M.; Figueiredo, C.; Leite, M.; Carlini, C.R. A New Role for Helicobacter pylori Urease: Contributions to Angiogenesis. Front. Microbiol. 2017, 8, 1883. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Stack, A.; Katzowitsch, E.; Aizawa, S.I.; Suerbaum, S.; Josenhans, C. Helicobacter pylori flagellins have very low intrinsic activity to stimulate human gastric epithelial cells via TLR5. Microbes Infect. 2003, 5, 1345–1356. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Yan, J.; Mao, Y.-F. Helicobacter pylori lipopolysaccharide: Biological activities in vitro and in vivo, pathological correlation to human chronic gastritis and peptic ulcer. World J. Gastroenterol. 2004, 10, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Lina, T.T.; Alzahrani, S.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Immune evasion strategies used by Helicobacter pylori. World J. Gastroenterol. 2014, 20, 12753–12766. [Google Scholar] [CrossRef] [PubMed]
- Andersen-Nissen, E.; Smith, K.D.; Strobe, K.L.; Barrett, S.L.R.; Cookson, B.T.; Logan, S.M.; Aderem, A. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc. Natl. Acad. Sci. USA 2005, 102, 9247. [Google Scholar] [CrossRef] [Green Version]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van der Vlist, M.; Geijtenbeek, T.B.H. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat. Immunol. 2009, 10, 1081–1088. [Google Scholar] [CrossRef]
- Tan, G.M.Y.; Looi, C.Y.; Fernandez, K.C.; Vadivelu, J.; Loke, M.F.; Wong, W.F. Suppression of cell division-associated genes by Helicobacter pylori attenuates proliferation of RAW264.7 monocytic macrophage cells. Sci. Rep. 2015, 5, 11046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramarao, N.; Meyer, T.F. Helicobacter pylori Resists Phagocytosis by Macrophages: Quantitative Assessment by Confocal Microscopy and Fluorescence-Activated Cell Sorting. Infect. Immun. 2001, 69, 2604–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lekmeechai, S.; Su, Y.-C.; Brant, M.; Alvarado-Kristensson, M.; Vallström, A.; Obi, I.; Arnqvist, A.; Riesbeck, K. Helicobacter pylori Outer Membrane Vesicles Protect the Pathogen From Reactive Oxygen Species of the Respiratory Burst. Front. Microbiol. 2018, 9, 1837. [Google Scholar] [CrossRef] [Green Version]
- Codolo, G.; Toffoletto, M.; Chemello, F.; Coletta, S.; Soler Teixidor, G.; Battaggia, G.; Munari, G.; Fassan, M.; Cagnin, S.; de Bernard, M. Helicobacter pylori Dampens HLA-II Expression on Macrophages via the Up-Regulation of miRNAs Targeting CIITA. Front. Immunol. 2020, 10, 2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, J.Y.; Zhang, M.; Miller, M.J.; Mills, J.C.; Wang, B.; Liu, M.; Eaton, K.A.; Zou, W.; Berndt, B.E.; Cole, T.S.; et al. Helicobacter pylori Immune Escape Is Mediated by Dendritic Cell–Induced Treg Skewing and Th17 Suppression in Mice. Gastroenterology 2010, 138, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lina, T.T.; Pinchuk, I.V.; House, J.; Yamaoka, Y.; Graham, D.Y.; Beswick, E.J.; Reyes, V.E. CagA-Dependent Downregulation of B7-H2 Expression on Gastric Mucosa and Inhibition of Th17 Responses during Helicobacter pylori Infection. J. Immunol. 2013, 191, 3838–3846. [Google Scholar] [CrossRef] [Green Version]
- Sarajlic, M.; Neuper, T.; Vetter, J.; Schaller, S.; Klicznik, M.M.; Gratz, I.K.; Wessler, S.; Posselt, G.; Horejs-Hoeck, J. H. pylori modulates DC functions via T4SS/TNFα/p38-dependent SOCS3 expression. Cell Commun. Signal. 2020, 18, 160. [Google Scholar] [CrossRef]
- Lv, Y.-p.; Cheng, P.; Zhang, J.-y.; Mao, F.-y.; Teng, Y.-s.; Liu, Y.-g.; Kong, H.; Wu, X.-l.; Hao, C.-j.; Han, B.; et al. Helicobacter pylori–induced matrix metallopeptidase-10 promotes gastric bacterial colonization and gastritis. Sci. Adv. 2019, 5, eaau6547. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rivera, C.; Bhatty, M.; Christie, P.J. Mechanism and function of type IV secretion during infection of the human host. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Frick-Cheng, A.E.; Pyburn, T.M.; Voss, B.J.; McDonald, W.H.; Ohi, M.D.; Cover, T.L. Molecular and Structural Analysis of the Helicobacter pylori Type IV Secretion System Core Complex. MBio 2016, 7, e02001-15. [Google Scholar] [CrossRef] [Green Version]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; König, W.; et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Hartig, R.; Delahay, R.M.; Rohde, M.; Brandt, S.; Conradi, J.; Takahashi, S.; Smolka, A.J.; Sewald, N.; Backert, S. A Small Fibronectin-mimicking Protein from Bacteria Induces Cell Spreading and Focal Adhesion Formation. J. Biol. Chem. 2010, 285, 23515–23526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishijima, N.; Suzuki, M.; Ashida, H.; Ichikawa, Y.; Kanegae, Y.; Saito, I.; Borén, T.; Haas, R.; Sasakawa, C.; Mimuro, H. BabA-mediated Adherence Is a Potentiator of the Helicobacter pylori Type IV Secretion System Activity. J. Biol. Chem. 2011, 286, 25256–25264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semper, R.P.; Vieth, M.; Gerhard, M.; Mejías-Luque, R. Helicobacter pylori Exploits the NLRC4 Inflammasome to Dampen Host Defenses. J. Immunol. 2019, 203, 2183. [Google Scholar] [CrossRef]
- Hacker, J.; Kaper, J.B. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 2000, 54, 641–679. [Google Scholar] [CrossRef] [Green Version]
- Abu-Taleb, A.M.F.; Abdelattef, R.S.; Abdel-Hady, A.A.; Omran, F.H.; El-korashi, L.A.; Abdel-aziz El-hady, H.; El-Gebaly, A.M. Prevalence of Helicobacter pylori cagA and iceA Genes and Their Association with Gastrointestinal Diseases. Int. J. Microbiol. 2018, 2018, 4809093. [Google Scholar] [CrossRef] [Green Version]
- Kamogawa-Schifter, Y.; Yamaoka, Y.; Uchida, T.; Beer, A.; Tribl, B.; Schöniger-Hekele, M.; Trauner, M.; Dolak, W. Prevalence of Helicobacter pylori and its CagA subtypes in gastric cancer and duodenal ulcer at an Austrian tertiary referral center over 25 years. PLoS ONE 2018, 13, e0197695. [Google Scholar] [CrossRef]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 Is a Key Mediator of Helicobacter pylori CagA Protein Activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef]
- Imai, S.; Ooki, T.; Murata-Kamiya, N.; Komura, D.; Tahmina, K.; Wu, W.; Takahashi-Kanemitsu, A.; Knight, C.T.; Kunita, A.; Suzuki, N.; et al. Helicobacter pylori CagA elicits BRCAness to induce genome instability that may underlie bacterial gastric carcinogenesis. Cell Host Microbe 2021, 29, 941–958.e910. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Feng, Y.; Hu, Y.; He, C.; Xie, C.; Ouyang, Y.; Artim, S.C.; Huang, D.; Zhu, Y.; Luo, Z.; et al. Helicobacter pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway. J. Exp. Clin. Cancer Res. 2018, 37, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, N.; Tay, A.C.Y.; Marshall, B.J.; Jain, U. Helicobacter pylori VacA, a distinct toxin exerts diverse functionalities in numerous cells: An overview. Helicobacter 2019, 24, e12544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bernard, M.; Burroni, D.; Papini, E.; Rappuoli, R.; Telford, J.; Montecucco, C. Identification of the Helicobacter pylori VacA Toxin Domain Active in the Cell Cytosol. Infect. Immun. 1998, 66, 6014–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, M.S.; Beckett, A.C.; Cover, T.L. Helicobacter pylori Vacuolating Toxin and Gastric Cancer. Toxins 2017, 9, 316. [Google Scholar] [CrossRef] [Green Version]
- Atherton, J.C.; Cao, P.; Peek, R.M.; Tummuru, M.K.R.; Blaser, M.J.; Cover, T.L. Mosaicism in Vacuolating Cytotoxin Alleles of Helicobacter pylori: Association of Specific vacA Types with Cytotoxin Production and Peptic Ulceration. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahiro, K.; Niidome, T.; Kimura, M.; Hatakeyama, T.; Aoyagi, H.; Kurazono, H.; Imagawa, K.-i.; Wada, A.; Moss, J.; Hirayama, T. Activation of Helicobacter pylori VacA Toxin by Alkaline or Acid Conditions Increases Its Binding to a 250-kDa Receptor Protein-tyrosine Phosphatase β. J. Biol. Chem. 1999, 274, 36693–36699. [Google Scholar] [CrossRef] [Green Version]
- de Bernard, M.; Papini, E.; de Filippis, V.; Gottardi, E.; Telford, J.; Manetti, R.; Fontana, A.; Rappuoli, R.; Montecucco, C. Low pH Activates the Vacuolating Toxin of Helicobacter pylori, Which Becomes Acid and Pepsin Resistant. J. Biol. Chem. 1995, 270, 23937–23940. [Google Scholar] [CrossRef] [Green Version]
- Ilver, D.; Barone, S.; Mercati, D.; Lupetti, P.; Telford, J.L. Helicobacter pyloritoxin VacA is transferred to host cells via a novel contact-dependent mechanism. Cell. Microbiol. 2004, 6, 167–174. [Google Scholar] [CrossRef]
- Montecucco, C.; Rappuoli, R. Living dangerously: How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2001, 2, 457. [Google Scholar] [CrossRef]
- Pelicic, V.; Reyrat, J.-M.; Sartori, L.; Pagliaccia, C.; Rappuoli, R.; Telford, J.L.; Montecucco, C.; Papini, E. Helicobacter pylori VacA cytotoxin associated with the bacteria increases epithelial permeability independently of its vacuolating activity. Microbiology 1999, 145, 2043–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, D.; Zambon, C.F.; Letley, D.P.; Stranges, A.; Marchet, A.; Rhead, J.L.; Schiavon, S.; Guariso, G.; Ceroti, M.; Nitti, D.; et al. Clinical Relevance of Helicobacter pylori cagA and vacA Gene Polymorphisms. Gastroenterology 2008, 135, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Azuma, T.; Ito, S.; Miyaji, H.; Hirai, M.; Yamazaki, Y.; Sato, F.; Kato, T.; Kohli, Y.; Kuriyama, M. Analysis and typing of the vacA gene from cagA-positive strains of Helicobacter pylori isolated in Japan. J. Clin. Microbiol. 1997, 35, 1710–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.J.; Lee, J.; Oh, S.J.; Yoon, M.S.; Jang, S.S.; Holland, R.L.; Reno, M.L.; Hamad, M.N.; Maeda, T.; Chung, H.J.; et al. Helicobacter pylori infection modulates host cell metabolism through VacA-dependent inhibition of mTORC1. Cell Host Microbe 2018, 23, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimian, G.; Sanei, M.H.; Shirzad, H.; Azadegan-Dehkordi, F.; Taghikhani, A.; Salimzadeh, L.; Hashemzadeh-Chaleshtori, M.; Rafieian-Kopaei, M.; Bagheri, N. Virulence factors of Helicobacter pylori vacA increase markedly gastric mucosal TGF-β1 mRNA expression in gastritis patients. Microb. Pathog. 2014, 67–68, 1–7. [Google Scholar] [CrossRef]
- Zhu, P.; Xue, J.; Zhang, Z.-J.; Jia, Y.-P.; Tong, Y.-N.; Han, D.; Li, Q.; Xiang, Y.; Mao, X.-H.; Tang, B. Helicobacter pylori VacA induces autophagic cell death in gastric epithelial cells via the endoplasmic reticulum stress pathway. Cell Death Dis. 2017, 8, 3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, M.; Arichi, T.; Nakazawa, T.; Berzofsky, J.A. Persistent infection by Helicobacter pylori down-modulates virus-specific CD8+ cytotoxic T cell response and prolongs viral infection. J. Infect. Dis. 1998, 177, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Elios, M.M.; Manghetti, M.; De Carli, M.; Costa, F.; Baldari, C.T.; Burroni, D.; Telford, J.L.; Romagnani, S.; Del Prete, G. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 1997, 158, 962–967. [Google Scholar]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori Vacuolating Cytotoxin Inhibits T Lymphocyte Activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef] [Green Version]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A Model for Gastric Cancer Epidemiology. Lancet 1975, 306, 58–60. [Google Scholar] [CrossRef]
- Correa, P. Is Gastric Cancer Preventable? Gut 2004, 53, 1217–1219. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Kuo, S.-H.; Chu, P.-Y.; Shan, Y.-S.; Tsai, C.-R.; Tsai, H.-J.; Chen, L.-T. The Epidemiology of Gastric Cancers in the Era of Helicobacter pylori Eradication: A Nationwide Cancer Registry-Based Study in Taiwan. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Mallepally, N.; Hammad, T.; Liu, Y.; Thrift, A.P.; El-Serag, H.B.; Tan, M.C. Prevalence of Helicobacter pylori Positive Non-cardia Gastric Adenocarcinoma Is Low and Decreasing in a US Population. Dig. Dis. Sci. 2020, 65, 2403–2411. [Google Scholar] [CrossRef]
- Duan, F.; Song, C.; Zhang, J.; Wang, P.; Ye, H.; Dai, L.; Zhang, J.; Wang, K. Evaluation of the Epidemiologic Efficacy of Eradicating Helicobacter pylori on Development of Gastric Cancer. Epidemiol. Rev. 2019, 41, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Fiori-Duarte, A.T.; Rodrigues, R.P.; Kitagawa, R.R.; Kawano, D.F. Insights into the Design of Inhibitors of the Urease Enzyme—A Major Target for the Treatment of Helicobacter pylori Infections. Curr. Med. Chem. 2020, 27, 3967–3982. [Google Scholar] [CrossRef] [PubMed]
- Debraekeleer, A.; Remaut, H. Future perspective for potential Helicobacter pylori eradication therapies. Future Microbiol. 2018, 13, 671–687. [Google Scholar] [CrossRef] [Green Version]
- Gollapalli, P.; Selvan G, T.; Manjunatha, H.; Shetty, P.; Kumari N, S. Genome-scale protein interaction network construction and topology analysis of functional hypothetical proteins in Helicobacter pylori divulges novel therapeutic targets. Microb. Pathog. 2021, 161, 105293. [Google Scholar] [CrossRef]
- Cheok, Y.Y.; Tan, G.M.Y.; Fernandez, K.C.; Chan, Y.T.; Lee, C.Y.Q.; Cheong, H.C.; Looi, C.Y.; Vadivelu, J.; Abdullah, S.; Wong, W.F. Podoplanin Drives Motility of Active Macrophage via Regulating Filamin C During Helicobacter pylori Infection. Front. Immunol. 2021, 12, 702156. [Google Scholar] [CrossRef]
- Desanti, G.; Saghir, A.; Naylor, A.; Kemble, S.; Falconer, J.; Wehmeyer, C.; Marshall, J.; Nakamura, K.; Goodall, M.; Navarro-Núñez, L.; et al. O014 Podoplanin (GP38), a marker of synovial inflammation, is an excellent therapeutic target in mouse collagen-induced arthritis. Ann. Rheum. Dis. 2018, 77, A7–A8. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Tang, B.; Lei, Y.; Yang, M.; Wang, S.; Hu, S.; Xie, Z.; Liu, Y.; Vlodavsky, I.; Yang, S. Helicobacter pylori-Induced Heparanase Promotes H. pylori Colonization and Gastritis. Front. Immunol. 2021, 12, 675747. [Google Scholar] [CrossRef]
- Chu, T.-H.; Huang, S.-T.; Yang, S.-F.; Li, C.-J.; Lin, H.-W.; Weng, B.-C.; Yang, S.-M.; Huang, S.-C.; Wu, J.-C.; Chang, Y.-C.; et al. Hepatoma-derived growth factor participates in Helicobacter Pylori-induced neutrophils recruitment, gastritis and gastric carcinogenesis. Oncogene 2019, 38, 6461–6477. [Google Scholar] [CrossRef]
- Abdi, E.; Latifi-Navid, S.; Abedi Sarvestani, F.; Esmailnejad, M.H. Emerging therapeutic targets for gastric cancer from a host-Helicobacter pylori interaction perspective. Expert Opin. Ther. Targets 2021, 25, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Sutton, P.; Boag, J.M. Status of vaccine research and development for Helicobacter pylori. Vaccine 2019, 37, 7295–7299. [Google Scholar] [CrossRef]
- Zeng, M.; Mao, X.H.; Li, J.X.; Tong, W.D.; Wang, B.; Zhang, Y.J.; Guo, G.; Zhao, Z.J.; Li, L.; Wu, D.L.; et al. Efficacy, safety, and immunogenicity of an oral recombinant Helicobacter pylori vaccine in children in China: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1457–1464. [Google Scholar] [CrossRef]
- Keikha, M.; Eslami, M.; Yousefi, B.; Ghasemian, A.; Karbalaei, M. Potential antigen candidates for subunit vaccine development against Helicobacter pylori infection. J. Cell. Physiol. 2019, 234, 21460–21470. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).