
Antiviral Immunity in SARS-CoV-2 Infection: From Protective to Deleterious Responses


 ,
,  ,
,  ,
,  ,
,  , ,
, , 

Abstract
:1. Introduction
2. Antiviral Immune Response in SARS-CoV-2 Infection
2.1. Antiviral Innate Response in SARS-CoV-2 Infection
2.2. Antiviral Adaptive Immune Response in SARS-CoV-2 Infection
2.2.1. Humoral-Mediated Immunity-Activation of B Cells
2.2.2. Cell-Mediated Immunity-Activation of T Cells
2.3. Deleterious Effects of the Immune Response in SARS-CoV-2 Infection
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092, Erratum in 2020, 183, 837. [Google Scholar] [CrossRef] [PubMed]
- Hasöksüz, M.; Kiliç, S.; Saraç, F. Coronaviruses and SARS-SARS-COV-2. Turk. J. Med. Sci. 2020, 21, 549–556. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.F. Lessons from the host defences of bats, a unique viral reservoir. Nature 2021, 589, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.-M. SARS-SRS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Understanding the complexities of SARS-CoV2 infection and its immunology: A road to immune-based therapeutics. Int. Immunopharmacol. 2020, 88, 106980. [Google Scholar] [CrossRef]
- Miao, Z.; Tidu, A.; Eriani, G.; Martin, F. Secondary structure of the SARS-SARS-CoV-2 5’-UTR. RNA Biol. 2021, 18, 447–456. [Google Scholar] [CrossRef]
- Beyer, D.K.; Forero, A. Mechanisms of Antiviral Immune Evasion of SARS-SARS-CoV-2. J. Mol. Biol. 2021, 167265. [Google Scholar] [CrossRef]
- Zhu, W.; Shyr, Z.; Lo, D.C.; Zheng, W. Viral Proteases as Targets for Coronavirus Disease 2019 Drug Development. J. Pharm. Exp. Ther. 2021, 378, 166–172. [Google Scholar] [CrossRef]
- Lotfi, M.; Rezaei, N. SARS-SARS-CoV-2: A comprehensive review from pathogenicity of the virus to clinical consequences. J. Med. Virol. 2020, 92, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Astuti, I.Y. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3, Erratum in 2020, 30, 3896. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and genomics of SARS-SARS-CoV-2: A review of the literature with the special focus on genetic diversity and SARS-SARS-CoV-2 genome detection. Genomics 2021, 113, 1221–1232. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.Á.; Filip, I.; Chowdhury, R.; Maranas, C.D.; Sorger, P.K.; AlQuraishi, M.; Rabadan, R. Recombination and lineage-specific mutations linked to the emergence of SARS-SARS-CoV-2. Genome Med. 2021, 13, 124. [Google Scholar] [CrossRef]
- Segreto, R.; Deigin, Y. The genetic structure of SARS-SARS-CoV-2 does not rule out a laboratory origin: SARS-SARS-COV-2 chimeric structure and furin cleavage site might be the result of genetic manipulation. Bioessays 2021, 43, e2000240. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Wu, C.; Zheng, M.; Yang, Y.; Gu, X.; Yang, K.; Li, M.; Liu, Y.; Zhang, Q.; Zhang, P.; Wang, Y.; et al. Furin: A Potential Therapeutic Target for COVID-19. iScience 2020, 23, 101642. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Aktas, B.; Aslim, B. Gut-lung axis and dysbiosis in COVID-19. Turk. J. Biol. 2020, 44, 265–272. [Google Scholar] [CrossRef]
- Zang, R.; Castro, M.F.G.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 mediate SARS-SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Pathogenesis of Virus Infections. In Fenner White’s Medical Virology; Academic Press: London, UK, 2017; pp. 77–104. [Google Scholar] [CrossRef]
- Kaslow, R.A. Epidemiology and control: Principles, practice and programs. In Viral Infections of Humans, 5th ed.; Kaslow, R.A., Stanberry, L.R., LeDuc, J.W., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Chapter 1. [Google Scholar]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Taki, K.; Gahlot, R.; Sharma, A.; Dhangar, K. A chronicle of SARS-SARS-CoV-2: Part-I-Epidemiology, diagnosis, prognosis, transmission and treatment. Sci. Total Environ. 2020, 734, 139278. [Google Scholar] [CrossRef] [PubMed]
- Shelhamer, M.C.; Wesson, P.D.; Solari, I.L.; Jensen, D.L.; Steele, W.A.; Dimitrov, V.G.; Kelly, J.D.; Aziz, S.; Gutierrez, V.P.; Vittinghoff, E.; et al. Prone Positioning in Moderate to Severe Acute Respiratory Distress Syndrome Due to COVID-19: A Cohort Study and Analysis of Physiology. J. Intensive Care Med. 2021, 36, 241–252. [Google Scholar] [CrossRef]
- Nathanson, N.; Ahmed, R.; Biron, C.A.; Brinton, M.A.; Gonzales-Scarano, F.; Griffin, D.E. Viral Pathogenesis and Immunity, 2nd ed.; Academic Press: London, UK, 2007. [Google Scholar]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009, 27, 313–338. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [Green Version]
- Vardhana, S.A.; Wolchok, J.D. The many faces of the anti-COVID immune response. J. Exp. Med. 2020, 217, e20200678. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.-C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Ni, L.; Ye, F.; Cheng, M.-L.; Feng, Y.; Deng, Y.-Q.; Zhao, H.; Wei, P.; Ge, J.; Gou, M.; Li, X.; et al. Detection of SARS-SARS-CoV-2 -Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity 2020, 52, 971–977. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majdoul, S.; Compton, A.A. Lessons in self-defence: Inhibition of virus entry by intrinsic immunity. Nat. Rev. Immunol. [CrossRef]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhang, E.; Gao, S.; Xiong, Y.; Lu, M. Toward a Functional Cure for Hepatitis B: The Rationale and Challenges for Therapeutic Targeting of the B Cell Immune Response. Front. Immunol. 2019, 10, 2308. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease. In B-Cell Activation by Armed Helper T Cells, 5th ed.; Garland Science: New York, NY, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK27142/ (accessed on 12 October 2021).
- Cano, R.L.E.; Lopera, H.D.E. Introduction to T and B lymphocytes. In Autoimmunity: From Bench to Bedside; Anaya, J.M., Shoenfeld, Y., Rojas-Villarraga, A., Eds.; El Rosario University Press: Bogota, Colombia, 2013; Chapter 5. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459471/ (accessed on 12 October 2021).
- Ten, B.T.; Wubbolts, R.; Stoorvogel, W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb. Perspect Biol. 2013, 5, a016873. [Google Scholar] [CrossRef] [Green Version]
- Ekkens, M.J.; Shedlock, D.J.; Jung, E.; Troy, A.; Pearce, E.L.; Shen, H.; Pearce, E.J. Th1 and Th2 cells help CD8 T-cell responses. Infect. Immun. 2007, 75, 2291–2296. [Google Scholar] [CrossRef] [Green Version]
- Cronkite, D.A.; Strutt, T.M. The Regulation of Inflammation by Innate and Adaptive Lymphocytes. J. Immunol. Res. 2018, 1467538. [Google Scholar] [CrossRef]
- Maloney, B.E.; Perera, K.D.; Saunders, D.R.D.; Shadipeni, N.; Fleming, S.D. Interactions of viruses and the humoral innate immune response. Clin. Immunol. 2020, 212, 108351. [Google Scholar] [CrossRef]
- Kohlmeier, J.E.; Woodland, D.L. Immunity to Respiratory Viruses. Annu. Rev. Immunol. 2009, 27, 61–82. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Izadi, S.; Callahan, M.; Deperalta, G.; Wecksler, A.T. Antibody-receptor interactions mediate antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2021, 297, 100826. [Google Scholar] [CrossRef]
- Klimpel, G.R. Immune Defenses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 50. Available online: https://www.ncbi.nlm.nih.gov/books/NBK8423/ (accessed on 4 December 2021).
- Palm, A.E.; Henry, C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front. Immunol. 2019, 10, 1787. [Google Scholar] [CrossRef] [Green Version]
- Welsh, R.M.; Selin, L.K.; Szomolanyi-Tsuda, E. Immunological memory to viral infections. Annu. Rev. Immunol. 2004, 22, 711–743. [Google Scholar] [CrossRef]
- Karthik, K.; Senthilkumar, T.M.A.; Udhayavel, S.; Raj, G.D. Role of antibody-dependent enhancement (ADE) in the virulence of SARS-SARS-CoV-2 and its mitigation strategies for the development of vaccines and immunotherapies to counter COVID-19. Hum. Vaccin. Immunother. 2020, 16, 3055–3060. [Google Scholar] [CrossRef]
- Burton, D. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2002, 2, 706–713. [Google Scholar] [CrossRef]
- Murchu, O.E.; Byrne, P.; Walsh, K.A.; Carty, P.G.; Connolly, M.; De Gascun, C.; Jordan, K.; Keoghan, M.; O’Brien, K.K.; O’Neill, M.; et al. Immune response following infection with SARS-SARS-CoV-2 and other coronaviruses: A rapid review. Rev. Med. Virol. 2021, 31, e2162. [Google Scholar] [CrossRef]
- Cancel, T.S.M.; Yoon, K.J. PathogenesisAntibody-Dependent Enhancement of Virus Infection and Disease. Viral Immunol. 2004, 16, 1. [Google Scholar]
- Sol, M.; Furuyama, W.; Marzi, A.; Carmody, A.B.; Maruyama, J.; Kuroda, M.; Miyamoto, H.; Nanbo, A.; Manzoor, R.; Yoshida, R.; et al. F cγ-receptor IIa-mediated Src Signaling Pathway Is Essential for the Antibody-Dependent Enhancement of Ebola Virus Infection. PLoS Pathog. 2016, 12, e1006139. [Google Scholar] [CrossRef]
- Maemura, T.; Kuroda, M.; Armbrust, T.; Yamayoshi, S.; Halfmann, P.J.; Kawaoka, Y. Antibody-Dependent Enhancement of SARS-SARS-CoV-2 Infection Is Mediated by the IgG Receptors FcγRIIA and FcγRIIIA but Does Not Contribute to Aberrant Cytokine Production by Macrophages. Mbio 2021, 12, e0198721. [Google Scholar] [CrossRef]
- Sun, P.; Kochel, T.J. The Battle between Infection and Host Immune Responses of Dengue Virus and Its Implication in Dengue Disease Pathogenesis. Sci. World J. 2013, 2013, 843469. [Google Scholar] [CrossRef] [Green Version]
- Kellam, P.; Barclay, W. The dynamics of humoral immune responses following SARS-SARS-CoV-2 infection and the potential for reinfection. J. Gen. Virol. 2020, 101, 791–797. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, Q.; Wang, H.; Liu, W.; Liao, X.; Su, Y.; Wang, X.; Yuan, J.; Li, T.; Li, J.; et al. Antibody Responses to SARS-SARS-CoV-2 in Patients with Novel Coronavirus Disease 2019. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 2027–2034. [Google Scholar] [CrossRef]
- Galipeau, Y.; Greig, M.; Liu, G.; Driedger, M.; Langlois, M.-A. Humoral Responses and Serological Assays in SARS-SARS-CoV-2 Infections. Front. Immunol. 2020, 11, 610688. [Google Scholar] [CrossRef]
- Ng, K.W.; Faulkner, N.; Cornish, G.H.; Rosa, A.; Harvey, R.; Hussain, S.; Ulferts, R.; Earl, C.; Wrobel, A.G.; Benton, D.J.; et al. Preexisting and de novo humoral immunity to SARS-SARS-CoV-2 in humans. Science 2020, 370, 1339–1343. [Google Scholar] [CrossRef]
- Post, N.; Eddy, D.; Huntley, C.; van Schalkwyk, M.C.I.; Shrotri, M.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. Antibody response to SARS-SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2020, 15, e0244126. [Google Scholar] [CrossRef]
- Maggi, E.; Canonica, G.W.; Moretta, L. COVID-19: Unanswered questions on immune response and pathogenesis. J. Allergy Clin. Immunol. 2020, 146, 18–22. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef]
- Nguyen-Contant, P.; Embong, A.K.; Kanagaiah, P.; Chaves, F.A.; Yang, H.; Branche, A.R.; Topham, D.J.; Sangster, M.Y. S Protein-Reactive IgG and Memory B Cell Production after Human SARS-SARS-CoV-2 Infection Includes Broad Reactivity to the S2 Subunit. MBio 2020, 11, e01991-20. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent antibody responses to SARS-SARS-CoV-2 in convalescent individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef]
- Seow, J.; Graham, C.; Merrick, B.; Acors, S.; Pickering, S.; Steel, K.J.A.; Hemmings, O.; O’Byrne, A.; Kouphou, N.; Galao, R.P.; et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-SARS-CoV-2 infection in humans. Nat. Microbiol. 2020, 5, 1598–1607. [Google Scholar] [CrossRef]
- Beaudoin-Bussières, G.; Laumaea, A.; Anand, S.P.; Prévost, J.; Gasser, R.; Goyette, G.; Medjahed, H.; Perreault, J.; Tremblay, T.; Lewin, A.; et al. Decline of Humoral Responses against SARS-SARS-CoV-2 Spike in Convalescent Individuals. MBio 2020, 11, e02590-20. [Google Scholar] [CrossRef] [PubMed]
- Isho, B.; Abe, K.T.; Zuo, M.; Jamal, A.J.; Rathod, B.; Wang, J.H.; Li, Z.; Chao, G.; Rojas, O.L.; Bang, Y.M.; et al. Persistence of serum and saliva antibody responses to SARS-SARS-CoV-2 spike antigens in COVID-19 patients. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Lan, L.; Xu, D.; Ye, G.; Xia, C.; Wang, S.; Li, Y.; Xu, H. Positive RT-PCR Test Results in Patients Recovered from COVID-19. JAMA 2020, 323, 1502–1503. [Google Scholar] [CrossRef] [Green Version]
- Carmo, A.; Pereira-Vaz, J.; Mota, V.; Mendes, A.; Morais, C.; da Silva, A.C.; Camilo, E.; Pinto, C.S.; Cunha, E.; Pereira, J.; et al. Clearance and persistence of SARS-SARS-CoV-2 RNA in patients with COVID-19. J. Med. Virol. 2020, 92, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Coronavirus reinfections: Three questions scientists are asking. Nature 2020, 585, 168–169. [Google Scholar] [CrossRef]
- Prado-Vivar, B.; Becerra-Wong, M.; Guadalupe, J.J.; Márquez, S.; Gutierrez, B.; Rojas-Silva, P.; Grunauer, M.; Trueba, G.; Barragán, V.; Cárdenas, P. A case of SARS-SARS-CoV-2 reinfection in Ecuador. Lancet Infect. Dis. 2021, 21, e142. [Google Scholar] [CrossRef]
- Gupta, V.; Bhoyar, R.C.; Jain, A.; Srivastava, S.; Upadhayay, R.; Imran, M.; Jolly, B.; Divakar, M.K.; Sharma, D.; Sehgal, P.; et al. Asymptomatic reinfection in two healthcare workers from India with genetically distinct SARS-SARS-CoV-2. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 73, e2823–e2825. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Bao, L.; Liu, J.; Xiao, C.; Liu, J.; Xue, J.; Lv, Q.; Qi, F.; Gao, H.; Yu, P.; et al. Primary exposure to SARS-SARS-CoV-2 protects against reinfection in rhesus macaques. Science 2020, 369, 818–823. [Google Scholar] [CrossRef]
- Hu, Z.; Zou, Q.; Su, B. Regulation of T cell immunity by cellular metabolism. Front. Med. 2018, 12, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon, M.S.; Engel, J.A.; Lee, H.J.; Haque, A. Development of circulating CD4(+) T-cell memory. Immunol. Cell Biol. 2019, 97, 617–624. [Google Scholar] [CrossRef]
- Nguyen, Q.P.; Deng, T.Z.; Witherden, D.A.; Goldrath, A.W. Origins of CD4(+) circulating and tissue-resident memory T-cells. Immunology 2019, 157, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Yewdell, J.W.; Haeryfar, S.M.M. Understanding presentation of viral antigens to CD8+ T cells in vivo: The key to rational vaccine design. Annu. Rev. Immunol. 2005, 23, 651–682. [Google Scholar] [CrossRef]
- Halle, S.; Halle, O.; Förster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-SARS-CoV-2 -reactive T cells in healthy donors and patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef]
- Mortaz, E.; Tabarsi, P.; Varahram, M.; Folkerts, G.; Adcock, I.M. The Immune Response and Immunopathology of COVID-19. Front. Immunol. 2020, 11, 2037. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Pierce, C.A.; Preston-Hurlburt, P.; Dai, Y.; Aschner, C.B.; Cheshenko, N.; Galen, B.; Garforth, S.J.; Herrera, N.G.; Jangra, R.K.; Morano, N.C.; et al. Immune responses to SARS-SARS-CoV-2 infection in hospitalized pediatric and adult patients. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiang, Y.; Du, H.; Wong, G.W.-K. SARS-SARS-CoV-2 infection in children-Understanding the immune responses and controlling the pandemic. Pediatr. Allergy Immunol. 2020, 31, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. Immunology of COVID-19. Environ. Microbiol. 2020, 22, 4895–4908. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-SARS-CoV-2 -specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.-B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Ko, J.-H.; Müller, M.A.; Seok, H.; Park, G.E.; Lee, J.Y.; Cho, S.Y.; Ha, Y.E.; Baek, J.Y.; Kim, S.H.; Kang, J.-M.; et al. Serologic responses of 42 MERS-coronavirus-infected patients according to the disease severity. Diagn. Microbiol. Infect. Dis. 2017, 89, 106–111. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Wang, P.-H.; Cheng, Y. Increasing Host Cellular Receptor—Angiotensin-Converting Enzyme 2 (ACE2) Expression by Coronavirus may Facilitate 2019-nCoV Infection. J. Med. Virol. 2020, 92, 2693–2701. [Google Scholar]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Toor, S.M.; Saleh, R.; Sasidharan Nair, V.; Taha, R.Z.; Elkord, E. T-cell responses and therapies against SARS-SARS-CoV-2 infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, M.-L.; Chien, C.-S.; Yarmishyn, A.A.; Yang, Y.-P.; Lai, W.-Y.; Luo, Y.-H.; Lin, Y.-T.; Chen, Y.-J.; Chang, P.C.; et al. Highlight of Immune Pathogenic Response and Hematopathologic Effect in SARS-CoV, MERS-CoV, and SARS-SARS-CoV-2 Infection. Front. Immunol. 2020, 11, 1022. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated with Drastically Elevated Interleukin 6 Level in Critically Ill Patients with Coronavirus Disease 2019. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.M.; Boyton, R.J. SARS-SARS-CoV-2 T cell immunity: Specificity, function, durability, and role in protection. Sci. Immunol. 2020, 5, eabd6160. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Qi, F.; Shi, K.; Li, Y.; Li, J.; Chen, Y.; Pan, J.; Zhou, T.; Lin, X.; Zhang, J.; et al. Thalidomide combined with low-dose short-term glucocorticoid in the treatment of critical Coronavirus Disease 2019. Clin. Transl Med. 2020, 10, e35. [Google Scholar] [CrossRef]
- Runfeng, L.; Yunlong, H.; Jicheng, H.; Weiqi, P.; Qinhai, M.; Yongxia, S.; Chufang, L.; Jin, Z.; Zhenhua, J.; Haiming, J.; et al. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2). Pharm. Res. 2020, 156, 104761. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. Coronavirus breakthrough: Dexamethasone is first drug shown to save lives. Nature 2020, 582, 469. [Google Scholar] [CrossRef]
- Marinella, M.A. Indomethacin and resveratrol as potential treatment adjuncts for SARS-CoV-2/COVID-19. Int. J. Clin. Pract. 2020, 74, e13535. [Google Scholar] [CrossRef]
- Yousefifard, M.; Zali, A.; Zarghi, A.; Madani Neishaboori, A.; Hosseini, M.; Safari, S. Non-steroidal anti-inflammatory drugs in management of COVID-19. A systematic review on current evidence. Int. J. Clin. Pract. 2020, 74, e13557. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Sayar, S.; Radmanesh, E.; Naghshi, S.; Mousaviasl, S.; Jelvay, S.; Ebrahimzadeh, M.; Mohammadi, A.; Abbasi, S.; Mobarak, S.; et al. Efficacy of naproxen in the management of patients hospitalized with COVID-19 infection: A randomized, double-blind, placebo-controlled, clinical trial. Diabetes Metab Syndr. 2021, 15, 102319. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, V.; Shannon, C.P.; Wei, X.S.; Xiang, X.; Wang, X.; Wang, Z.H.; Tebbutt, S.J.; Kollmann, T.R.; Fish, E.N. Interferon-α2b Treatment for COVID-19. Front. Immunol. 2020, 11, 1060. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D.; et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe 2020, 28, 455–464.e2. [Google Scholar] [CrossRef]
- Kapur, S.; Bonk, M.E. Rilonacept (arcalyst), an interleukin-1 trap for the treatment of cryopyrin-associated periodic syndromes. Pharm. Ther. 2009, 34, 138–141. [Google Scholar]
- Sánchez-Fernández, A.; Skouras, D.B.; Dinarello, C.A.; López-Vales, R. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front. Immunol. 2019, 10, 2578. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Huang, C.L.; Fei, L.; Xu, W.; Li, W.X.; Xie, X.D.; Li, Q.; Chen, L. Efficacy Evaluation of Thymosin Alpha 1 in Non-severe Patients With COVID-19: A Retrospective Cohort Study Based on Propensity Score Matching. Front. Med. 2021, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Zhu, C.; Liu, L.; Qi, T.; Shen, Y.; Zhang, Y.; Xu, L.; Li, T.; Qian, Z.; et al. Thymosin Alpha-1 Has no Beneficial Effect on Restoring CD4+ and CD8+ T Lymphocyte Counts in COVID-19 Patients. Front. Immunol. 2021, 12, 2172. [Google Scholar] [CrossRef] [PubMed]
- Sagawa, T.; Inoue, K.I.; Takano, H. Use of protease inhibitors for the prevention of COVID-19. Prev Med. 2020, 141, 106280. [Google Scholar] [CrossRef]
- Sciascia, S.; Apra, F.; Baffa, A.; Baldovino, S.; Boaro, D.; Boero, R.; Bonora, S.; Calcagno, A.; Cecchi, I.; Cinnirella, G.; et al. Pilot prospective open, single-arm multicentre study on off-label use of tocilizumab in patients with severe COVID-19. Clin. Exp. Rheumatol. 2020, 38, 529–532. [Google Scholar]
- Salama, C.; Han, J.; Yau, L.; Reiss, W.G.; Kramer, B.; Neidhart, J.D.; Criner, G.J.; Kaplan-Lewis, E.; Baden, R.; Pandit, L.; et al. Tocilizumab in Patients Hospitalized with Covid-19 Pneumonia. N. Engl. J. Med. 2021, 384, 20–30. [Google Scholar] [CrossRef]
- Roschewski, M.; Lionakis, M.S.; Sharman, J.P.; Roswarski, J.; Goy, A.; Monticelli, M.A.; Roshon, M.; Wrzesinski, S.H.; Desai, J.V.; Zarakas, M.A.; et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 2020, 5, eabd0110. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Tassan Din, C.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.M.; Bezie, Y.; Laplanche, S.; et al. Anakinra for severe forms of COVID-19: A cohort study. Lancet Rheumatol. 2020, 2, e393–e400. [Google Scholar] [CrossRef]
- Blech, M.; Peter, D.; Fischer, P.; Bauer, M.M.; Hafner, M.; Zeeb, M.; Nar, H. One target-two different binding modes: Structural insights into gevokizumab and canakinumab interactions to interleukin-1β. J. Mol. Biol. 2013, 425, 94–111. [Google Scholar] [CrossRef]
- Zheng, Z.H.; Zeng, X.; Nie, X.Y.; Cheng, Y.J.; Liu, J.; Lin, X.X.; Yao, H.; Ji, C.-C.; Chen, X.-M.; Jun, F.; et al. Interleukin-1 blockade treatment decreasing cardiovascular risk. Clin. Cardiol. 2019, 42, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Keewan, E.; Beg, S.; Naser, S.A. Anti-TNF-α agents Modulate SARS-CoV-2 Receptors and Increase the Risk of Infection Through Notch-1 Signaling. Front. Immunol. 2021, 12, 1662. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchié, A.; Wang, T.S.; Lee, E.; Cremer, P.C.; Carey, B.; Rajendram, P.; Hudock, K.M.; Korbee, L.; Van Tassell, B.W.; et al. Targeting GM-CSF in COVID-19 Pneumonia: Rationale and Strategies. Front. Immunol. 2020, 11, 1625. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, H.M.; Ragsdale, C.E.; Gale, R.P.; Lyman, G.H. Sargramostim (rhu GM-CSF) as Cancer Therapy (Systematic Review) and An Immunomodulator. A Drug Before Its Time? Front. Immunol. 2021, 17, 12. [Google Scholar] [CrossRef]
- A Phase II/III Study of Sargramostim-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04642950 (accessed on 5 December 2021).
- Sargramostim Use in COVID-19 to Recover Patient Health-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04707664 (accessed on 5 December 2021).
- Wright, D.J.M. Prevention of the cytokine storm in COVID-19. Lancet Infect. Dis. 2021, 21, 25–26. [Google Scholar] [CrossRef]
- Golchin, A.; Seyedjafari, E.; Ardeshirylajimi, A. Mesenchymal Stem Cell Therapy for COVID-19: Present or Future. Stem Cell Rev. Rep. 2020, 16, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Rezakhani, L.; Kelishadrokhi, A.F.; Soleimanizadeh, A.; Rahmati, S. Mesenchymal stem cell (MSC)-derived exosomes as a cell-free therapy for patients Infected with COVID-19: Real opportunities and range of promises. Chem. Phys. Lipids 2021, 1, 234. [Google Scholar] [CrossRef]
- Mesenchymal Stem Cells in Patients Diagnosed with COVID-19-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04611256 (accessed on 5 December 2021).
- Supady, A.; Weber, E.; Rieder, M.; Lother, A.; Niklaus, T.; Zahn, T.; Frech, F.; Müller, S.; Kuhl, M.; Benk, C.; et al. Cytokine adsorption in patients with severe COVID-19 pneumonia requiring extracorporeal membrane oxygenation (CYCOV): A single centre, open-label, randomised, controlled trial. Lancet Respir Med. 2021, 9, 755–762. [Google Scholar] [CrossRef]
- Zuccari, S.; Damiani, E.; Domizi, R.; Scorcella, C.; D’Arezzo, M.; Carsetti, A.; Pantanetti, S.; Vannicola, S.; Casarotta, E.; Ranghino, A.; et al. Changes in Cytokines, Haemodynamics and Microcirculation in Patients with Sepsis/Septic Shock Undergoing Continuous Renal Replacement Therapy and Blood Purification with CytoSorb. Blood Purif. 2020, 49, 107–113. [Google Scholar] [CrossRef]
- Turnquist, C.; Ryan, B.M.; Horikawa, I.; Harris, B.T.; Harris, C.C. Cytokine Storms in Cancer and COVID-19. Cancer Cell 2020, 38, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, R.; Zaffina, S.; Piano Mortari, E.; Terreri, S.; Corrente, F.; Capponi, C.; Palomba, P.; Mirabella, M.; Cascioli, S.; Palange, P.; et al. Different Innate and Adaptive Immune Responses to SARS-SARS-CoV-2 Infection of Asymptomatic, Mild, and Severe Cases. Front. Immunol. 2020, 11, 610300. [Google Scholar] [CrossRef]
- Seyed, H.; Safiabadi, T.; Jason, J.; LeBlanc, Z.; Sadiq, O.; Damilola, O.; Carolina, C. Tools and Techniques for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)/COVID-19 Detection. Clin. Microbiol. Rev. 2020, 34, e0022820. [Google Scholar]
- Leisman, D.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis and comparison with other inflammatory syndromes. Lancet 2020, 8, 2020. [Google Scholar] [CrossRef]
- Schaefer, E.J.; Dulipsingh, L.; Comite, F.; Jimison, J.; Grajower, M.M.; Lebowitz, N.E.; Lang, M.; Geller, A.S.; Diffenderfer, M.R.; He, L.; et al. Corona Virus Disease-19 serology, inflammatory markers, hospitalizations, case finding and aging. PLoS ONE 2021, 16, e0252818. [Google Scholar] [CrossRef] [PubMed]
- Iliescu, F.S.; Ionescu, A.M.; Gogianu, L.; Simion, M.; Dediu, V.; Chifiriuc, M.C.; Pircalabioru, G.G.; Iliescu, C. Point-of-Care Testing-the Key in the Battle against SARS-CoV-2 Pandemic. Micromachines 2021, 12, 1464. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef]
- Clay, C.; Donart, N.; Fomukong, N.; Knight, J.B.; Lei, W.; Price, L.; Hahn, F.; Van Westrienen, J.; Harrod, K.S. Primary severe acute respiratory syndrome coronavirus infection limits replication but not lung inflammation upon homologous rechallenge. J. Virol. 2012, 86, 4234–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aid, M.; Busman-Sahay, K.; Vidal, S.J.; Maliga, Z.; Bondoc, S.; Starke, C.; Terry, M.; Jacobson, C.A.; Wrijil, L.; Ducat, S.; et al. Vascular Disease and Thrombosis in SARS-SARS-CoV-2 -Infected Rhesus Macaques. Cell 2020, 183, 1354–1366.e13. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. Found: Genes that sway the course of the coronavirus. Science 2020, 370, 275–276. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Gorbunova, V.; Seluanov, A.; Kennedy, B.K. The World Goes Bats: Living Longer and Tolerating Viruses. Cell Metab. 2020, 32, 31–43. [Google Scholar] [CrossRef]
- Bargehr, J.; Rericha, P.; Petchey, A.; Colzani, M.; Moule, G.; Malgapo, M.C.; Rassl, D.; Tarkin, J.; Mellor, G.; Sampaziotis, F.; et al. Cardiovascular ACE2 receptor expression in patients undergoing heart transplantation. ESC Hear. Fail. 2021, 8, 4119–4129. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Tucker, N.R.; Chaffin, M.; Bedi, K.C., Jr.; Papangeli, I.; Akkad, A.-D.; Arduini, A.; Hayat, S.; Eraslan, G.; Muus, C.; Bhattacharyya, R.P.; et al. Myocyte-Specific Upregulation of ACE2 in Cardiovascular Disease: Implications for SARS-SARS-CoV-2 –Mediated Myocarditis. Circulation 2020, 142, 708. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celardo, I.; Pace, L.; Cifaldi, L.; Gaudio, C.; Barnaba, V. The immune system view of the coronavirus SARS-SARS-CoV-2. Biol. Direct 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.-C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1–7 axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, N.; Kuo, H.-H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef]
- Takahashi, T.; Ellingson, M.K.; Wong, P.; Israelow, B.; Lucas, C.; Klein, J.; Silva, J.; Mao, T.; Oh, J.E.; Tokuyama, M.; et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 2020, 588, 315–320. [Google Scholar] [CrossRef]
- Ibarrondo, F.J.; Fulcher, J.A.; Goodman-Meza, D.; Elliott, J.; Hofmann, C.; Hausner, M.A.; Ferbas, K.G.; Tobin, N.H.; Aldrovandi, G.M.; Yang, O.O. Rapid Decay of Anti-SARS-SARS-CoV-2 Antibodies in Persons with Mild Covid-19. N. Engl. J. Med. 2020, 383, 1085–1087. [Google Scholar] [CrossRef]
- Long, Q.-X.; Tang, X.-J.; Shi, Q.-L.; Li, Q.; Deng, H.-J.; Yuan, J.; Hu, J.-L.; Xu, W.; Zhang, Y.; Lv, F.-J.; et al. Clinical and immunological assessment of asymptomatic SARS-SARS-CoV-2 infections. Nat. Med. 2020, 26, 1200–1204. [Google Scholar] [CrossRef]
- Yu, H.-Q.; Sun, B.-Q.; Fang, Z.-F.; Zhao, J.-C.; Liu, X.-Y.; Li, Y.-M.; Sun, X.-Z.; Liang, H.-F.; Zhong, B.; Huang, Z.-F.; et al. Distinct features of SARS-CoV-2 -specific IgA response in COVID-19 patients. Eur. Respir. J. 2020, 56, 2001526. [Google Scholar] [CrossRef]
- Newell, K.L.; Clemmer, D.C.; Cox, J.B.; Ayode, Y.I.; Zoccoli-Rodriguez, V.; Taylor, H.E.; Endy, T.P.; Wilmore, J.R.; Winslow, G.M. Switched and unswitched memory B cells detected during SARS-SARS-CoV-2 convalescence correlate with limited symptom duration. PLoS ONE 2021, 16, e0244855. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Møller, R.; Panis, M.; Sachs, D.; Albrecht, R.A.; tenOever, B.R. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, J.; Legge, K.; Perlman, S. Age-related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Investig. 2011, 121, 4921–4930. [Google Scholar] [CrossRef]
- García, L.F. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-SARS-CoV-2 -Reactive CD4+ T Cells in COVID-19. Cell 2020, 183, 1340–1353.e16. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef] [PubMed]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef] [PubMed]

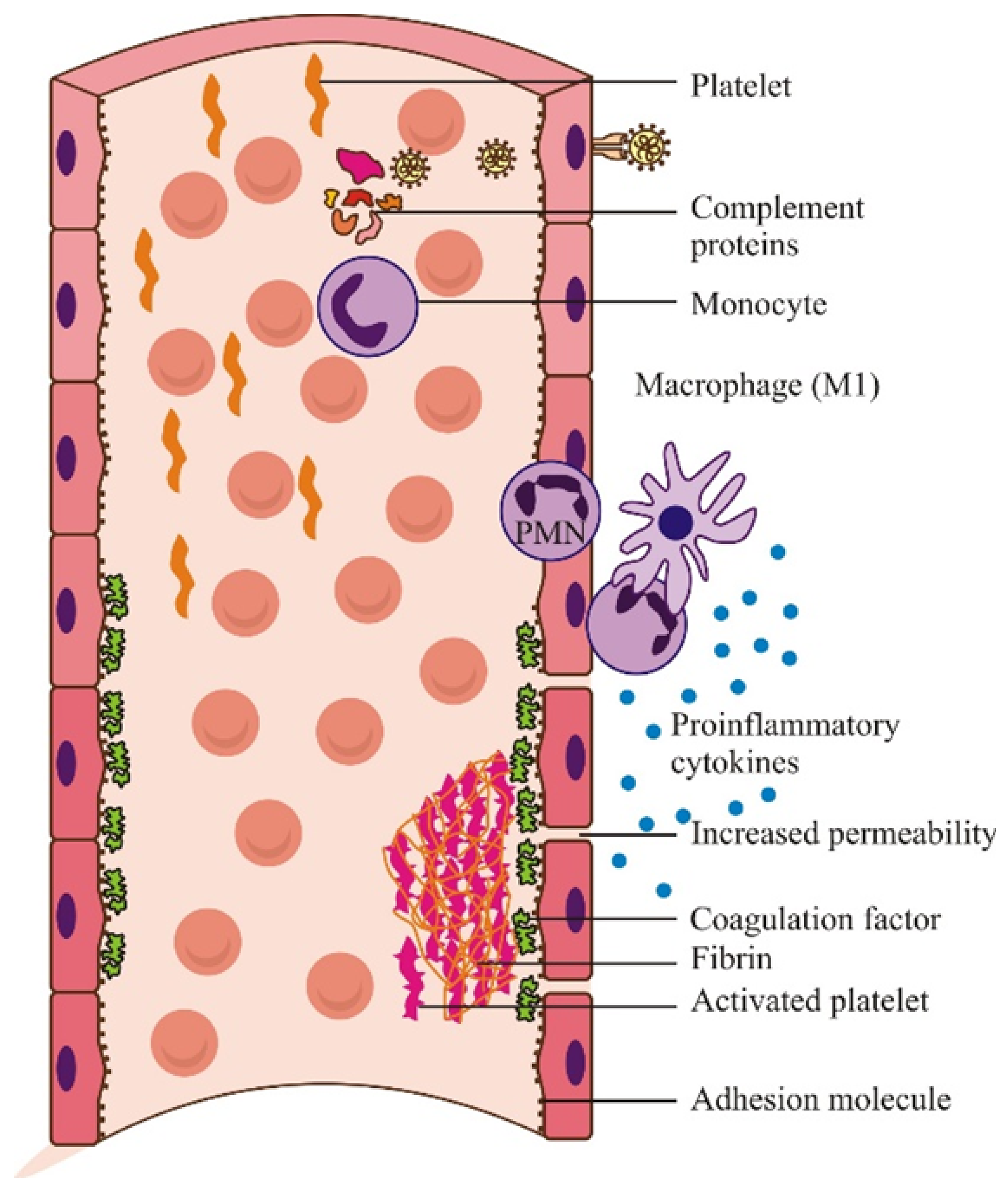

{kind=link}
{kind=link}
| Entry Gate | Local Defense Mechanisms | |
|---|---|---|
| Cutaneous tissue | Desiccation Acid pH Temperature variations Desquamation of epithelial cells Resident microbiota at this level (competition for adhesion and colonization sites) Hair follicles and sebaceous glands Subcutaneous lymphoid tissue | |
| Mucosa The mucin layer traps pathogens Submucosa lymphoid tissue | Conjunctival mucosa | The blinking reflex periodically removes pathogens that have reached this level Conjunctival secretion (sIgA) |
| Nasopharynx | Resident microbiota Secretions (sIgA, phagocytes) | |
| Upper respiratory tract | Mucus Ciliary movements | |
| Lung | Macrophages | |
| Gastro-intestinal and genito-urinary tract | Periodic desquamation sIgA Complement Resident microbiota Low pH Proteolytic enzymes Low/High fluid velocity Low fluid velocity | |
| Antiviral Immunity Components | Innate | Adaptive |
|---|---|---|
| Molecular factors | Intrinsic antiviral substances, interferons, complement, pro-inflammatory cytokines | Specific antibodies (IgG, IgM, IgA) |
| Cells | Natural killer (NK), phagocytes, dendritic cells (DCs) | T and B cells |
| Primary infection | + | + |
| Secondary Infection | + | +++ |
| Immunological memory | − | + |
| Drug Class | Target/Pathway | Name | Effect |
|---|---|---|---|
| Immunosuppressive/ Immunomodulatory drugs | Anti-inflammatory, anti-angiogenic, and anti-fibrotic immune modulator | Thalidomide | Combined with GC, prevents SARS-COV2 pneumonia [108] |
| Anti-inflammatory | Lianhuaqingwen | Reduces TNF-α, IL-6, CXCL10, MCP-1 levels [109] | |
| Anti-inflammatory, prevents cellular autophagy | Chloroquine/ Hydroxychloroquine | Recovers the levels of NK cells and CD8+ T cells [110] | |
| Anti-inflammatory | Corticosteroids (dexamethasone) | Reduces death in severe cases [111] RECOVERY Collaborative Group, 2021 | |
| Anti-inflammatory, immunosuppressive | Naproxen Indomethacin | Reduces viral load, suppresses RNA synthesis [112,113,114] | |
| Anti-inflammatory, antiviral | IFN-α2b | Reduces duration of detectable virus, reduces circulating levels IL-6, CRP [115] | |
| IFN-α2b with or without umifenovir | NCT04354259, NCT04343976, NCT04344600, NCT04388709 [116] | ||
| Anti-inflammatory, NLRP3 inflammasome inhibitor | Rilonacept Inzomelid, Somalix, Dapansutrile (small molecule NLRP3 inhibitors) | [117,118,119] | |
| Activating and regulating immune cells Restores CD4+ and CD8+ T Cells Counts | Thymosin α1 | Shorten viral RNA shedding duration and hospital stay [120,121] | |
| JAK signaling inhibitors | Baricitinib Fedratinib Ruxolitinib | Blockage of virus entry and the attenuation of host excessive inflammatory response NCT04970719 NCT04477993 | |
| Serine protease inhibitors | Nafamostat mesylate Camostat mesylate | inhibitors of complement pathways and broad-spectrum anti-inflammatory agents [122] | |
| Modulation of the sphingosine-1-phosphate receptor 1 pathway | Fingolimod | sphingosine-1-phosphate receptor modulator, which sequesters lymphocytes in lymph nodes, preventing them from contributing to an autoimmune reaction NCT04280588 (https://clinicaltrials.gov/ct2/show/NCT04280588 (accessed on 5 December 2021) | |
| Monoclonal antibodies | Antagonist of the IL-6 receptor | Tocilizumab and sarilumab | Reduce the cytokine storm [123,124] |
| Recombinant mAb that binds to both soluble and membrane-bound IL-6 | Siltuximab | NCT04330638 | |
| Bruton’s tyrosine kinase (BTK) inhibitor- effects on the signaling of TLRs, IL-1R, CD19, BCR, CXCR4, and Fcγ-R1 | Acalabrutinib Ibrutinib Acalabrutinib | [125] Clinical trials: NCT04375397 NCT04439006 NCT04564040 NCT04380688 | |
| Recombinant antagonist of the IL-1 receptor | Anakinra, Gevokizumab, Canakinumab | Reduce death and hospitalization [119,126,127,128,129,130] | |
| Anti-TNF-α antibody | infliximab, adalimumab, certolizumab pegol | Reduce the cytokine storm [131,132] ChiCTR2000030089 | |
| Anti-GM-CSF monoclonal antibody | Lenzilumab Gimsilumab Namilumab | Reduce the cytokine storm [133] | |
| Rhu GM-CSF | Sargramostim | [133,134,135,136] | |
| Partial opioid agonist | Meptazinol | Reduce the cytokine storm [137] | |
| MSC-based therapy | MSC-derived exosomes (MSC-Exo) | Trials ongoing [138,139,140] | |
| Blood purification therapy | Hemodialysis, hemofiltration, plasma exchange, and hemoperfusion | Cytosorb | Reduce the cytokine storm [141,142] |
| Parameter | Change | Specimen Type | COVID Status | Biosafety | Reference |
|---|---|---|---|---|---|
| Lymphocyte count | Lymphopenia | Blood | Increased severity | Clinical laboratory | [145] |
| Neutrophile count | Neutropenia | Blood | Increased severity | Clinical laboratory | [145] |
| ALT, AST, LDH, CRP, Ferritin | Elevated | serum | Increased severity | Clinical laboratory | [145] |
| IL-6 | increased | serum | Critical illness | Clinical laboratory | [145] |
| D-dimer, lymphopenia | Elevated levels | serum | Risk of death | Clinical laboratory | [145] |
| IL-8 | Moderately increased | serum | Moderate severity | Clinical laboratory | [146] |
| IFN γ | Not elevated | serum | All forms | Clinical laboratory | [146] |
| SARS-CoV 2 IgG antibody | Increased | serum | Chronic COVID Increased in positive older subjects | Clinical laboratory/biosafety level 2 | [147,148] |
| SARS-CoV 2 IgM antibody | Increased | serum | Persistent COVID 10 symptoms and disease | Clinical Laboratory/biosafety level 2 | [147,148] |
| Neutralizing antibodies | Increased | serum | Indicate immunity; monitor vaccine effectiveness | Clinical laboratory/biosafety level 3 | [148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihaescu, G.; Chifiriuc, M.C.; Vrancianu, C.O.; Constantin, M.; Filip, R.; Popescu, M.R.; Burlibasa, L.; Nicoara, A.C.; Bolocan, A.; Iliescu, C.; et al. Antiviral Immunity in SARS-CoV-2 Infection: From Protective to Deleterious Responses. Microorganisms 2021, 9, 2578. https://doi.org/10.3390/microorganisms9122578
Mihaescu G, Chifiriuc MC, Vrancianu CO, Constantin M, Filip R, Popescu MR, Burlibasa L, Nicoara AC, Bolocan A, Iliescu C, et al. Antiviral Immunity in SARS-CoV-2 Infection: From Protective to Deleterious Responses. Microorganisms. 2021; 9(12):2578. https://doi.org/10.3390/microorganisms9122578
Chicago/Turabian StyleMihaescu, Grigore, Mariana Carmen Chifiriuc, Corneliu Ovidiu Vrancianu, Marian Constantin, Roxana Filip, Mihaela Roxana Popescu, Liliana Burlibasa, Anca Cecilia Nicoara, Alexandra Bolocan, Ciprian Iliescu, and et al. 2021. "Antiviral Immunity in SARS-CoV-2 Infection: From Protective to Deleterious Responses" Microorganisms 9, no. 12: 2578. https://doi.org/10.3390/microorganisms9122578
APA StyleMihaescu, G., Chifiriuc, M. C., Vrancianu, C. O., Constantin, M., Filip, R., Popescu, M. R., Burlibasa, L., Nicoara, A. C., Bolocan, A., Iliescu, C., & Gradisteanu Pircalabioru, G. (2021). Antiviral Immunity in SARS-CoV-2 Infection: From Protective to Deleterious Responses. Microorganisms, 9(12), 2578. https://doi.org/10.3390/microorganisms9122578