
Variation among Metschnikowia pulcherrima Isolates for Genetic Modification and Homologous Recombination


Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Media
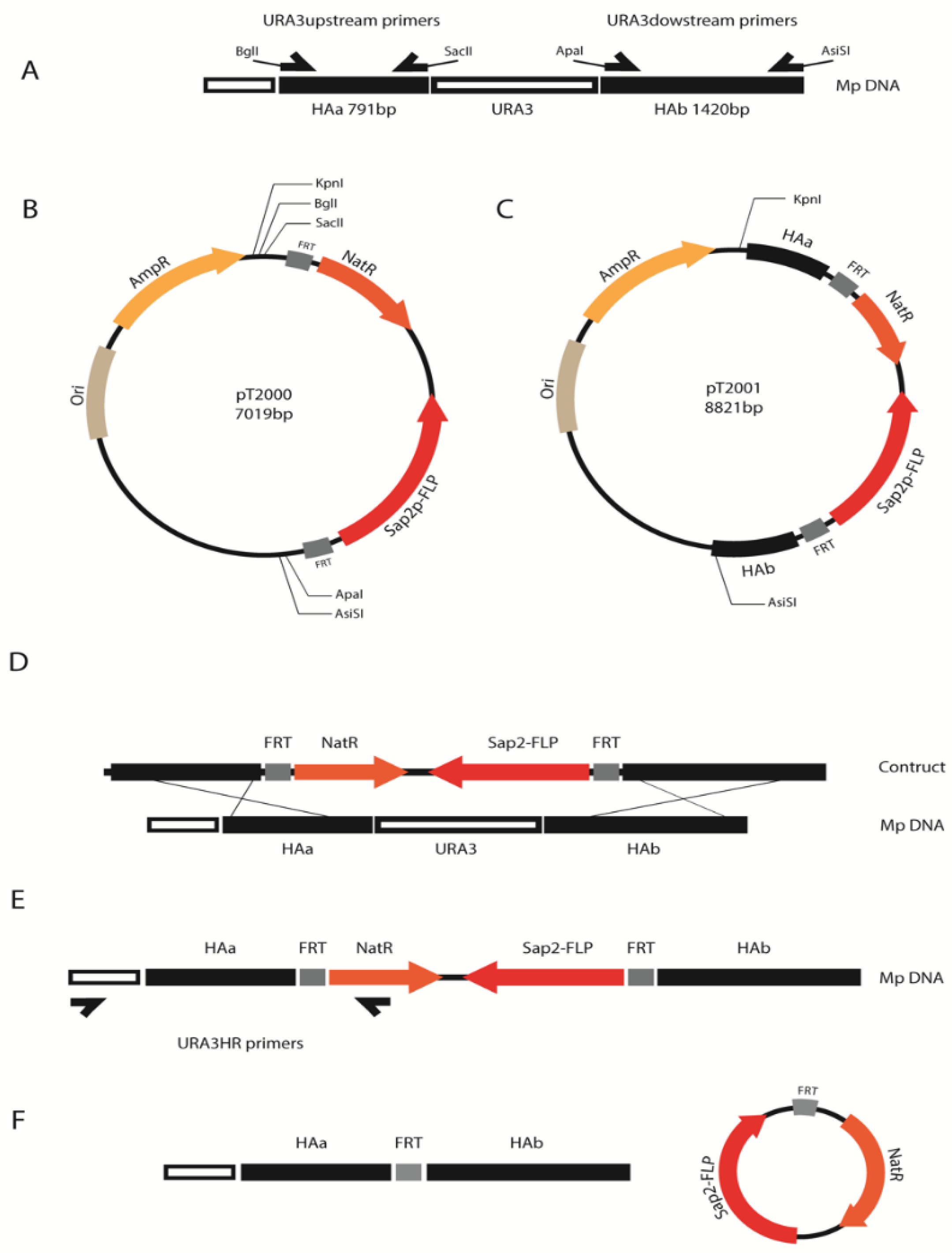
2.2. Generation of the URA3 Deletion Construct
2.3. Yeast Transformation
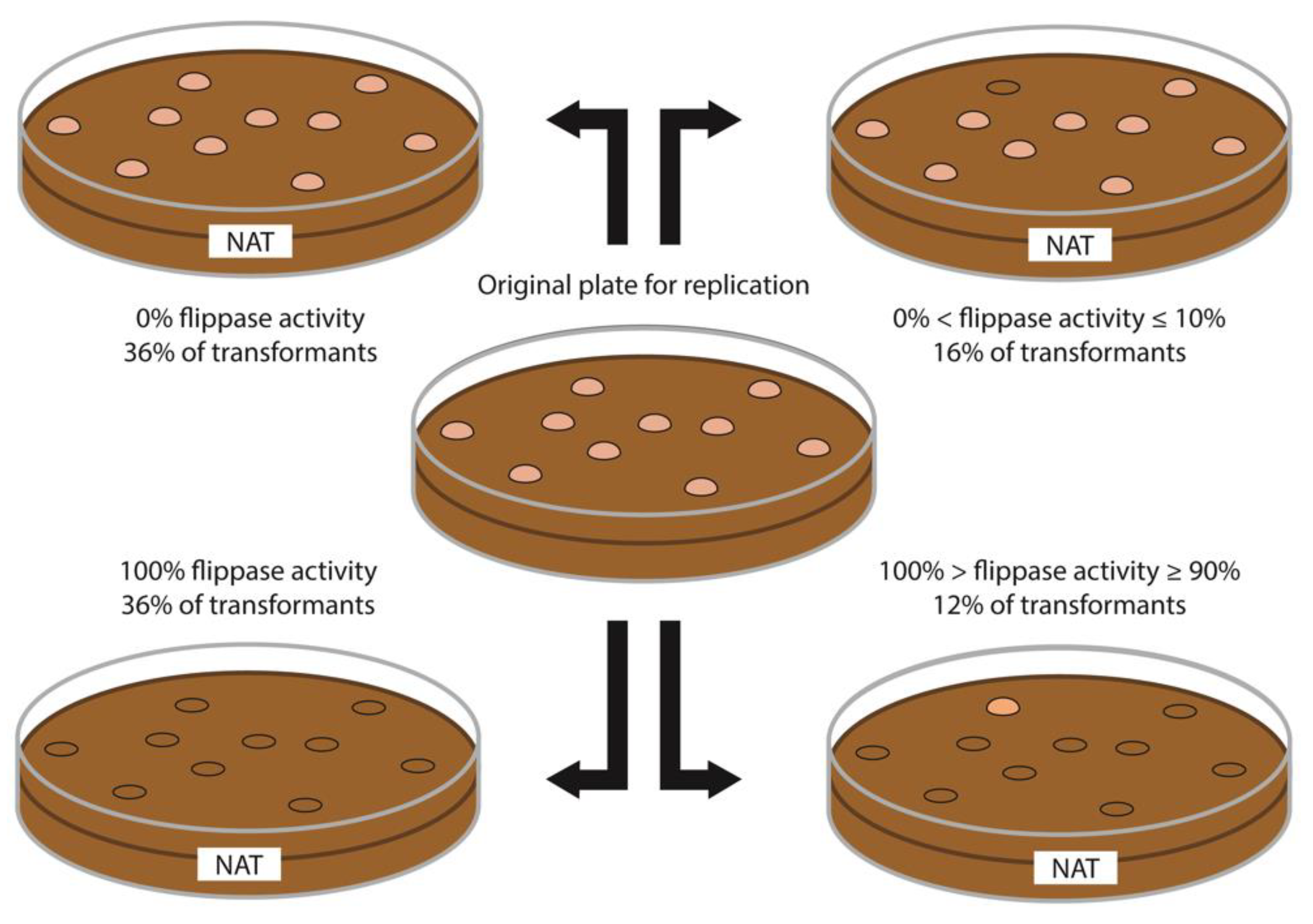
2.4. Transformants Screening
2.5. Heat Shock Evaluation
2.6. Drug Treatments to Increase HR
2.7. Selection Marker Recycling
3. Results
3.1. Strain Level Variation in HR and Response to Heat Shock
3.2. Chemical Suppression of NHEJ in a Strain with Low but Detectable HR
3.3. Marker Recycling via FLP in a Diploid Strain
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Papon, N.; Courdavault, V.; Clastre, M. Biotechnological Potential of the Fungal CTG Clade Species in the Synthetic Biology Era. Trends Biotechnol. 2014, 32, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.; Haridas, S.; Wolfe, K.H.; Lopes, M.R.; Todd, C.; Göker, M. Comparative Genomics of Biotechnologically Important Yeasts. Proc. Natl. Acad. Sci. USA 2016, 113, 9882–9887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensels, J.; Verstrepen, K.J. Taming Wild Yeast: Potential of Conventional and Nonconventional Yeasts in Industrial Fermentations. Annu. Rev. Microbiol. 2014, 68, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Mannazzu, I.; Landolfo, S.; da Silva, T.L.; Buzzini, P. Red Yeasts and Carotenoid Production: Outlining a Future for Non-Conventional Yeasts of Biotechnological Interest. World J. Microbiol. Biotechnol. 2015, 31, 1665–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masneuf-pomarede, I.; Bely, M.; Marullo, P.; Albertin, W. The Genetics of Non-Conventional Wine Yeasts: Current Knowledge and Future Challenges. Front. Microbiol. 2016, 6, 1563. [Google Scholar] [CrossRef] [PubMed]
- Nel, S.; Labuschagne, M.; Albertyn, J. Advances in Gene Expression in Non-Conventional Yeasts. In Yeast Biotechnology: Diversity and Applications; Satyanarayana, T., Kunze, G., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 369–403. ISBN 978-1-4020-8292-4. [Google Scholar]
- Qiao, K.; Abidi, S.H.I.; Liu, H.; Zhang, H.; Chakraborty, S.; Watson, N.; Ajikumar, P.K.; Stephanopoulos, G. Engineering Lipid Overproduction in the Oleaginous Yeast Yarrowia Lipolytica. Metab. Eng. 2015, 29, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Spohner, S.C.; Schaum, V.; Quitmann, H.; Czermak, P. Kluyveromyces Lactis: An Emerging Tool in Biotechnology. J. Biotechnol. 2016, 222, 104–116. [Google Scholar] [CrossRef]
- Mortimer, R.K.; Johnston, J.R. Genealogy of Principal Strains of the Yeast Genetic Stock Center. Genetics 1986, 113, 35–43. [Google Scholar] [CrossRef]
- Kawai, S.; Hashimoto, W.; Murata, K. Transformation of Saccharomyces Cerevisiae and Other Fungi: Methods and Possible Underlying Mechanism. Bioeng. Bugs 2010, 1, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Gietz, R.D.; Schiestl, R.H. High-Efficiency Yeast Transformation Using the LiAc/SS Carrier DNA/PEG Method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef]
- Ito, H.; Fukuda, Y.; Murata, K.; Kimura, A. Transformation of Intact Yeast Cells Treated with Alkali Cations. J. Bacteriol. 1983, 153, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, K.A.; Vazquez, S.; Alper, H.S. High-Efficiency Transformation of Yarrowia Lipolytica Using Electroporation. FEMS Yeast Res. 2018, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Wendland, J. An Improved Transformation Protocol for the Human Fungal Pathogen Candida Albicans. Curr. Genet. 2003, 42, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Klinner, U.; Schafer, B. Genetic Aspects of Targeted Insertion Mutagenesis in Yeasts. FEMS Microbiol. Rev. 2004, 28, 201–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, P.; Gao, J.; Zhou, Y. CRISPR-Mediated Genome Editing in Non-Conventional Yeasts for Biotechnological Applications. Microb. Cell Factories 2019, 18, 63. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-Strand Break Repair Pathway Choice. Cell Res. 2007, 18, 134. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Sun, H.; Li, P.; He, N.; Zhu, T.; Li, Y. Enhancement of the Gene Targeting Efficiency of Non-Conventional Yeasts by Increasing Genetic Redundancy. PLoS ONE 2013, 8, e57952. [Google Scholar] [CrossRef]
- Kretzschmar, A.; Otto, C.; Holz, M.; Werner, S.; Hubner, L.; Barth, G. Increased Homologous Integration Frequency in Yarrowia Lipolytica Strains Defective in Non-Homologous End-Joining. Curr. Genet. 2013, 59, 63–72. [Google Scholar] [CrossRef]
- Foureau, E.; Courdavault, V.; Rojas, L.F.; Dutilleul, C.; Simkin, A.J.; Crèche, J.; Atehortùa, L.; Giglioli-Guivarc’h, N.; Clastre, M.; Papon, N. Efficient Gene Targeting in a Candida Guilliermondii Non-Homologous End-Joining Pathway-Deficient Strain. Biotechnol. Lett. 2013, 35, 1035–1043. [Google Scholar] [CrossRef]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A Dynamic Enzyme in a Versatile DSB Repair Pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattah, F.J.; Lichter, N.F.; Fattah, K.R.; Oh, S.; Hendrickson, E.A. Ku70, an Essential Gene, Modulates the Frequency of RAAV-Mediated Gene Targeting in Human Somatic Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, G.; Rio, D. DNA Double-Strand-Break Sensitivity, DNA Replication, and Cell Cycle Arrest Phenotypes of Ku-Deficient Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 867–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arras, S.D.M.; Fraser, J.A. Chemical Inhibitors of Non-Homologous End Joining Increase Targeted Construct Integration in Cryptococcus Neoformans. PLoS ONE 2016, 11, e0163049. [Google Scholar] [CrossRef] [PubMed]
- Tsakraklides, V.; Brevnova, E.; Stephanopoulos, G.; Shaw, A.J. Improved Gene Targeting through Cell Cycle Synchronization. PLoS ONE 2015, 10, e0133434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronostajski, R.M.; Sadowski, P.D. The FLP Recombinase of the Saccharomyces Cerevisiae 2 Microns Plasmid Attaches Covalently to DNA via a Phosphotyrosyl Linkage. Mol. Cell. Biol. 1985, 5, 3274–3279. [Google Scholar] [CrossRef] [Green Version]
- Morschhauser, J.; Michel, S.; Staib, P. Sequential Gene Disruption in Candida Albicans by FLP-Mediated Site-Specific Recombination. Mol. Microbiol. 1999, 32, 547–556. [Google Scholar] [CrossRef]
- Yamada, Y.; Maeda, M.; Alshahni, M.M.; Monod, M.; Staib, P.; Yamada, T. Flippase (FLP) Recombinase-Mediated Marker Recycling in the Dermatophyte Arthroderma Vanbreuseghemii. Microbiology 2014, 160, 2122–2135. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Santomauro, F.; Budarin, V.L.; Whiffin, F.; Abeln, F.; Chantasuban, T.; Gore-Lloyd, D.; Henk, D.; Scott, R.J.; Clark, J.; et al. The Additive Free Microwave Hydrolysis of Lignocellulosic Biomass for Fermentation to High Value Products. J. Clean. Prod. 2018, 198, 776–784. [Google Scholar] [CrossRef]
- Santamauro, F.; Whiffin, F.M.; Scott, R.J.; Chuck, C.J. Low-Cost Lipid Production by an Oleaginous Yeast Cultured in Non-Sterile Conditions Using Model Waste Resources. Biotechnol. Biofuels 2014, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sipiczki, M. Metschnikowia Pulcherrima and Related Pulcherrimin-Producing Yeasts: Fuzzy Species Boundaries and Complex Antimicrobial Antagonism. Microorganisms 2020, 8, 1029. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, B.; Lachance, M.-A.; Herrera, C.M. Phylogenetic Analysis of the Angiosperm-Floricolous Insect–Yeast Association: Have Yeast and Angiosperm Lineages Co-Diversified? Mol. Phylogenetics Evol. 2013, 68, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.I.; Miller, M.W. Sporulation in Candida Pulcherrima, Candida Reukaufii and Chlamydozyma Species: Their Relationships with Metschnikowia. Mycologia 1968, 60, 663–685. [Google Scholar] [CrossRef]
- Oro, L.; Ciani, M.; Comitini, F. Antimicrobial Activity of Metschnikowia Pulcherrima on Wine Yeasts. J. Appl. Microbiol. 2014, 116, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Sipiczki, M. Metschnikowia Strains Isolated from Botrytized Grapes Antagonize Fungal and Bacterial Growth by Iron Depletion. Appl. Environ. Microbiol. 2006, 72, 6716–6724. [Google Scholar] [CrossRef] [Green Version]
- Gore-Lloyd, D.; Sumann, I.; Brachmann, A.O.; Schneeberger, K.; Ortiz-Merino, R.A.; Moreno-Beltrán, M.; Schläfli, M.; Kirner, P.; Kron, A.S.; Rueda-Mejia, M.P.; et al. Snf2 Controls Pulcherriminic Acid Biosynthesis and Antifungal Activity of the Biocontrol Yeast Metschnikowia Pulcherrima. Mol. Microbiol. 2019, 112, 317–332. [Google Scholar] [CrossRef] [Green Version]
- Hershkovitz, V.; Sela, N.; Taha-Salaime, L.; Liu, J.; Rafael, G.; Kessler, C.; Aly, R.; Levy, M.; Wisniewski, M.; Droby, S. De-Novo Assembly and Characterization of the Transcriptome of Metschnikowia Fructicola Reveals Differences in Gene Expression Following Interaction with Penicillium Digitatum and Grapefruit Peel. BMC Genom. 2013, 14, 168. [Google Scholar] [CrossRef] [Green Version]
- Kurtzman, C.P.; Droby, S. Metschnikowia Fructicola, a New Ascosporic Yeast with Potential for Biocontrol of Postharvest Fruit Rots. Syst. Appl. Microbiol. 2001, 24, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Morata, A.; Loira, I.; Escott, C.; del Fresno, J.M.; Bañuelos, M.A.; Suárez-Lepe, J.A. Applications of Metschnikowia Pulcherrima in Wine Biotechnology. Fermentation 2019, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Contreras, A.; Curtin, C.; Varela, C. Yeast Population Dynamics Reveal a Potential ‘Collaboration’ between Metschnikowia Pulcherrima and Saccharomyces Uvarum for the Production of Reduced Alcohol Wines during Shiraz Fermentation. Appl. Microbiol. Biotechnol. 2014, 99, 1885–1895. [Google Scholar] [CrossRef]
- Chantasuban, T.; Santomauro, F.; Gore-Lloyd, D.; Parsons, S.; Henk, D.; Scott, R.J.; Chuck, C. Elevated Production of the Aromatic Fragrance Molecule, 2-Phenylethanol, Using Metschnikowia Pulcherrima through Both de Novo and Ex Novo Conversion in Batch and Continuous Modes. J. Chem. Technol. Biotechnol. 2018, 93, 2118–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeln, F.; Hicks, R.H.; Auta, H.; Moreno-Beltrán, M.; Longanesi, L.; Henk, D.A.; Chuck, C.J. Semi-Continuous Pilot-Scale Microbial Oil Production with Metschnikowia Pulcherrima on Starch Hydrolysate. Biotechnol. Biofuels 2020, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Abeln, F.; Fan, J.; Budarin, V.L.; Briers, H.; Parsons, S.; Allen, M.J.; Henk, D.A.; Clark, J.; Chuck, C.J. Lipid Production through the Single-Step Microwave Hydrolysis of Macroalgae Using the Oleaginous Yeast Metschnikowia Pulcherrima. Algal Res. 2019, 38, 101411. [Google Scholar] [CrossRef] [Green Version]
- Janisiewicz, W.J.; Tworkoski, T.J.; Kurtzman, C.P. Biocontrol Potential of Metchnikowia Pulcherrima Strains Against Blue Mold of Apple. Phytopathology 2001, 91, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnár, O.; Prillinger, H. Analysis of Yeast Isolates Related to Metschnikowia Pulcherrima Using the Partial Sequences of the Large Subunit RDNA and the Actin Gene; Description of Metschnikowia Andauensis Sp. Nov. Syst. Appl. Microbiol. 2005, 28, 717–726. [Google Scholar] [CrossRef]
- Nigro, F.; Sialer, M.M.F.; Gallitelli, D.S. Transformation of Metschnikowia Pulcherrima 320, Biocontrol Agent of Storage Rot, with The Green Fluorescent Protein Gene. J. Plant Pathol. 1999, 81, 205–208. [Google Scholar]
- Hicks, R.H.; Chuck, C.J.; Scott, R.J.; Leak, D.J.; Henk, D.A. Comparison of Nile Red and Cell Size Analysis for High-Throughput Lipid Estimation Within Oleaginous Yeast. Eur. J. Lipid Sci. Technol. 2019, 121, 1800355. [Google Scholar] [CrossRef] [Green Version]
- Hicks, R.H.; Sze, Y.; Chuck, C.J.; Henk, D.A. Enhanced Inhibitor Tolerance and Increased Lipid Productivity through Adaptive Laboratory Evolution in the Oleaginous Yeast Metshnikowia Pulcherrima. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kwolek-Mirek, M.; Zadrag-Tecza, R. Comparison of Methods Used for Assessing the Viability and Vitality of Yeast Cells. FEMS Yeast Res. 2014, 14, 1068–1079. [Google Scholar] [CrossRef]
- Guyot, S.; Gervais, P.; Young, M.; Winckler, P.; Dumont, J.; Davey, H.M. Surviving the Heat: Heterogeneity of Response in Saccharomyces Cerevisiae Provides Insight into Thermal Damage to the Membrane. Environ. Microbiol. 2015, 17, 2982–2992. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Conley, A.J.; Penttilä, M.; Jäntti, J.; Wang, H.H.; Church, G.M. Yeast Oligo-Mediated Genome Engineering (YOGE). ACS Synth. Biol. 2013, 2, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.G.; David, F.; Siewers, V.; Nielsen, J. Engineering Lipid Droplet Assembly Mechanisms for Improved Triacylglycerol Accumulation in Saccharomyces Cerevisiae. FEMS Yeast Res. 2018, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-L.; Li, B.-Z.; Zhang, W.-Z.; Song, K.; Qi, H.; Dai, J.-B.; Yuan, Y.-J. Genome-Wide Landscape of Position Effects on Heterogeneous Gene Expression in Saccharomyces Cerevisiae. Biotechnol. Biofuels 2017, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; Nicolas, A. Clustering of Meiotic Double-Strand Breaks on Yeast Chromosome III. Proc. Natl. Acad. Sci. USA 1997, 94, 5213–5218. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Strain ID | Source | Substrate | Notes | Country of Origin |
|---|---|---|---|---|
| 3047 | NCYC 3047 | Fruit of Phoenix dactylifera | Prototype strain | Egypt |
| 2580 | NCYC 2580 | Unknown | 20–25% lipid [48] | Unknown |
| FS | UBFCC 20131 | Fruit of Vitis vinifera | Grown in raceway pond [31] | UK |
| 4x3 | NCYC 4331 | Derived from 2580 | Inhibitor tolerant [43,49] | UK |
| DH5 | UBFCC 20145 | Fruit of Rubus sp. | 10–15% lipid [48] | UK |
| ICS48 | UBFCC 201546 | Fruit of Rubus sp. | 5–10% lipid [48] | UK |
| ICS46 | UBFCC 201548 | Fruit of Rubus sp. | 10–15% lipid [48] | UK |
| ICS1 | UBFCC 20151 | Fruit of Rubus sp. | Grown at scale [44] | UK |
| F3 | UBFCC 2016F3 | Derived from 2580 | Formic acid tolerant [49] | UK |
| DH10 | UBFCC 201410 | Fruit of Rubus sp. | Growth on algae [44] | UK |
| Primer Name | Sequence | Tm (°C) | Elongation Time (min) |
|---|---|---|---|
| BgIIURA3upstream_FW | attaagatctGTATTCACCGATAGATAGGC | 55 | 1 |
| SacIIURA3upstream_RV | cataccgcggACATGGTCACTCTAGCGGGC | 55 | 1 |
| ApaIURA3downstream_FW | gcatgggcccTAAAAGTTGTGTTTGAGCGTCGTC | 55 | 1 |
| AsiSIURA3downstream_RV | tgacgcgatcgcTCAGATGAACCTCCAGAGCCA | 55 | 1 |
| 5977HRScreen_FW | ACCTGACGTCCCGCCCATCGCGCTTTGACTACATG | 55 | 1 |
| NatHRScreen_RV | TCTCTCAAAGTGAAACCATCACCAGTAGC | 60 | 1 |
| BgIIURA3upstream_FW | gcatgggcccTAAAAGTTGTGTTTGAGCGTCGTC | 60 | 1 |
| Strain | Colonies Screened | % Survival in Additional Nat | % Colonies with HR | %Colonies with HR Surviving | Mean % Survived and HS | SD of Colonies Surviving HS |
|---|---|---|---|---|---|---|
| 3047 | 100 | 36 | 1 | 2.77 | 11,67 | 6.66 |
| 2580 | 100 | 16 | 2 | 12.5 | 42 | 5.20 |
| FS | 100 | 16 | 0 | 0 | 26.33 | 19.04 |
| 4x3 | 100 | 16 | 0 | 0 | 19.67 | 1.53 |
| DH5 | 100 | 13 | 0 | 0 | 29.67 | 7.57 |
| ICS48 | 100 | 11 | 0 | 0 | 63.33 | 13.80 |
| ICS46 | 100 | 10 | 0 | 0 | 34.33 | 5.86 |
| ICS1 | 100 | 8 | 0 | 0 | 40.33 | 9.29 |
| F3 | 30 | 33.66 | 0 | 0 | 19.67 | 15.57 |
| DH10 | 20 | 20 | 0 | 0 | 17.67 | 6.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Beltrán, M.; Gore-Lloyd, D.; Chuck, C.; Henk, D. Variation among Metschnikowia pulcherrima Isolates for Genetic Modification and Homologous Recombination. Microorganisms 2021, 9, 290. https://doi.org/10.3390/microorganisms9020290
Moreno-Beltrán M, Gore-Lloyd D, Chuck C, Henk D. Variation among Metschnikowia pulcherrima Isolates for Genetic Modification and Homologous Recombination. Microorganisms. 2021; 9(2):290. https://doi.org/10.3390/microorganisms9020290
Chicago/Turabian StyleMoreno-Beltrán, Mauro, Deborah Gore-Lloyd, Christopher Chuck, and Daniel Henk. 2021. "Variation among Metschnikowia pulcherrima Isolates for Genetic Modification and Homologous Recombination" Microorganisms 9, no. 2: 290. https://doi.org/10.3390/microorganisms9020290