
What Is the Best Lens? Comparing the Resolution Power of Genome-Derived Markers and Standard Barcodes

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Sequences
2.2. Capturing Markers from Genomes
2.3. Alignment and Data Analysis
2.4. Inter and Intra-Group Distances Analysis
2.5. Calculation of MeTRe
3. Results
3.1. Experimental Design
3.2. Distance Analysis among Saccharomyces Species
3.3. Distance Analysis among the Pathogenic Candida Species
3.4. Proposal of Mean Taxonomic Resolution (MeTRe) as a Novel Metric to Determine Marker Efficiency
3.5. Single-Copy Markers from Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dujon, B.; Sherman, D.; Fischer, G.; Durrens, P.; Casaregola, S.; Lafontaine, I.; de Montigny, J.; Marck, C.; Neuveglise, C.; Talla, E.; et al. Genome Evolution in Yeasts. Nature 2004, 430, 35–44. Available online: http://www.nature.com/nature/journal/v430/n6995/suppinfo/nature02579_S1.html (accessed on 1 October 2020). [CrossRef]
- Cardinali, G.; Corte, L.; Robert, V. Next Generation Sequencing: Problems and opportunities for next generation studies of microbial communities in food and food industry. Curr. Opin. Food Sci. 2017, 17, 62–67. [Google Scholar] [CrossRef]
- Borneman, A.R.; Pretorius, I.S. Genomic insights into the Saccharomyces sensu stricto complex. Genetics 2015, 199, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leducq, J.-B.; Nielly-Thibault, L.; Charron, G.; Eberlein, C.; Verta, J.-P.; Samani, P.; Sylvester, K.; Hittinger, C.T.; Bell, G.; Landry, C.R. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nat. Microbiol. 2016, 1, 1–10. [Google Scholar] [CrossRef]
- Louis, E.J. Population genomics and speciation in yeasts. Fungal Biol. Rev. 2011, 25, 136–142. [Google Scholar]
- Bruns, T.D.; White, T.J.; Taylor, J.W. Fungal molecular systematics. Annu. Rev. Ecol. Syst. 1991, 22, 525–564. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Skarshewski, A.; Parks, D.H. Genome-based microbial taxonomy coming of age. Cold Spring Harb. Perspect. Biol. 2016, 8, a018085. [Google Scholar] [CrossRef] [Green Version]
- Kurtzman, C.P.; Robnett, C.J. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Van Leeuwenhoek 1998, 73, 331–371. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J. Relationships among genera of the Saccharomycotina (Ascomycota) from multigene phylogenetic analysis of type species. FEMS Yeast Res. 2013, 13, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. Taxonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Yurkov, A.; Guerreiro, M.A.; Sharma, L.; Carvalho, C.; Fonseca, Á. Multigene assessment of the species boundaries and sexual status of the basidiomycetous yeasts Cryptococcus flavescens and C. terrestris (Tremellales). PLoS ONE 2015, 10, e0120400. [Google Scholar] [CrossRef] [PubMed]
- Steenkamp, E.T.; Wingfield, M.J.; McTaggart, A.R.; Wingfield, B.D. Fungal species and their boundaries matter—Definitions, mechanisms and practical implications. Fungal Biol. Rev. 2018, 32, 104–116. [Google Scholar] [CrossRef]
- Pigliucci, M. Species as family resemblance concepts: The (dis-) solution of the species problem? BioEssays 2003, 25, 596–602. [Google Scholar] [CrossRef] [Green Version]
- Lucking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; Groenewald, M.; Szöke, S.; Cardinali, G.; Eberhardt, U.; Stielow, B.; de Vries, M.; Verkley, G.J.M.; Crous, P.W.; Boekhout, T.; et al. DNA barcoding analysis of more than 9000 yeast isolates contributes to quantitative thresholds for yeast species and genera delimitation. Stud. Mycol. 2016, 85, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, G. Measure of species variability for a microbial taxonomy based on the relative resemblance. Riv. Biol. 2003, 96, 271–291. [Google Scholar]
- Irinyi, L.; Serena, C.; Garcia-Hermoso, D.; Arabatzis, M.; Desnos-Ollivier, M.; Vu, D.; Cardinali, G.; Arthur, I.; Normand, A.-C.; Giraldo, A. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—The quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med. Mycol. 2015, 53, myv008. [Google Scholar] [CrossRef]
- Schoch, C.L.; Robbertse, B.; Robert, V.; Vu, D.; Cardinali, G.; Irinyi, L.; Meyer, W.; Nilsson, R.H.; Hughes, K.; Miller, A.N.; et al. Finding needles in haystacks: Linking scientific names, reference specimens and molecular data for Fungi. Database J. Biol. Datrabase Curation 2014, 2014, bau061. [Google Scholar] [CrossRef]
- Gong, J.; Dong, J.; Liu, X.; Massana, R. Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 2013, 164, 369–379. [Google Scholar] [CrossRef]
- Li, W.; Sun, H.; Deng, Y.; Zhang, A.; Chen, H. The heterogeneity of the rDNA-ITS sequence and its phylogeny in Rhizoctonia cerealis, the cause of sharp eyespot in wheat. Curr. Genet. 2014, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weider, L.J.; Elser, J.J.; Crease, T.J.; Mateos, M.; Cotner, J.B.; Markow, T.A. The functional significance of ribosomal (r) DNA variation: Impacts on the evolutionary ecology of organisms. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 219–242. [Google Scholar] [CrossRef] [Green Version]
- West, C.; James, S.A.; Davey, R.P.; Dicks, J.; Roberts, I.N. Ribosomal DNA sequence heterogeneity reflects intraspecies phylogenies and predicts genome structure in two contrasting yeast species. Syst. Biol. 2014, 63, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, K.; Steenkamp, E.T.; Coetzee, M.P.; Wingfield, M.J.; Wingfield, B.D. Concerted evolution in the ribosomal RNA cistron. PLoS ONE 2013, 8, e59355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Rooney, A.P. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005, 39, 121. [Google Scholar] [CrossRef] [Green Version]
- Colabella, C.; Corte, L.; Roscini, L.; Bassetti, M.; Tascini, C.; Mellor, J.C.; Meyer, W.; Robert, V.; Vu, D.; Cardinali, G. NGS barcode sequencing in taxonomy and diagnostics, an application in “Candida” pathogenic yeasts with a metagenomic perspectiv. IMA Fungus 2018, 9, 91–105. [Google Scholar] [CrossRef]
- Roscini, L.; Tristezza, M.; Corte, L.; Colabella, C.; Perrotta, C.; Rampino, P.; Robert, V.; Vu, D.; Cardinali, G.; Grieco, F. Early Ongoing Speciation of Ogataea uvarum Sp. Nov. Within the Grape Ecosystem Revealed by the Internal Variability Among the rDNA Operon Repeats. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Stielow, J.; Lévesque, C.; Seifert, K.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.; Verkley, G.; Groenewald, M.; Chaduli, D. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Pers. Mol. Phylogeny Evol. Fungi 2015, 35, 242–263. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- The R Development Core Team. R: A Language and Environment for Statistical Computing; The R Development Core Team: Vienna, Austria, 2013. [Google Scholar]
- Schoch, C.L. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Tavanti, A.; Davidson, A.D.; Gow, N.A.; Maiden, M.C.; Odds, F.C. Candida orthopsilosis and Candida metapsilosis spp. nov. to replace Candida parapsilosis groups II and III. J. Clin. Microbiol. 2005, 43, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papon, N.; Courdavault, V.; Clastre, M.; Bennett, R.J. Emerging and emerged pathogenic Candida species: Beyond the Candida albicans paradigm. PLoS Pathog. 2013, 9, e1003550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonielli, L.; Robert, V.; Corte, L.; Roscini, L.; Ceppitelli, R.; Cardinali, G. Centrality of Objects in a Multidimensional Space and its Effects on Distance-Based Biological Classifications. Open Appl. Inform. J. 2011, 5, 11–19. [Google Scholar]
- Hennig, W. Phylogenetic systematics. Annu. Rev. Entomol. 1965, 10, 97–116. [Google Scholar] [CrossRef]
- Galtier, N. Delineating species in the speciation continuum: A proposal. Evol. Appl. 2019, 12, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, J.M.; Puerta-Fernandez, E.; Santana, M.M.; Rekadwad, B. On a Non-Discrete Concept of Prokaryotic Species. Microorganisms 2020, 8, 1723. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Utturkar, S.M.; Klingeman, D.M.; Hurt Jr, R.A.; Brown, S.D. A case study into microbial genome assembly gap sequences and finishing strategies. Front. Microbiol. 2017, 8, 1272. [Google Scholar] [CrossRef]
- Libkind, D.; Cadez, N.; Opulente, D.A.; Langdon, Q.K.; Rosa, C.A.; Sampaio, J.P.; Goncalves, P.; Hittinger, C.T.; Lachance, M.A. Towards yeast taxogenomics: Lessons from novel species descriptions based on complete genome sequences. FEMS Yeast Res. 2020, 20. [Google Scholar] [CrossRef]
- Matute, D.R.; Sepulveda, V.E. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef]







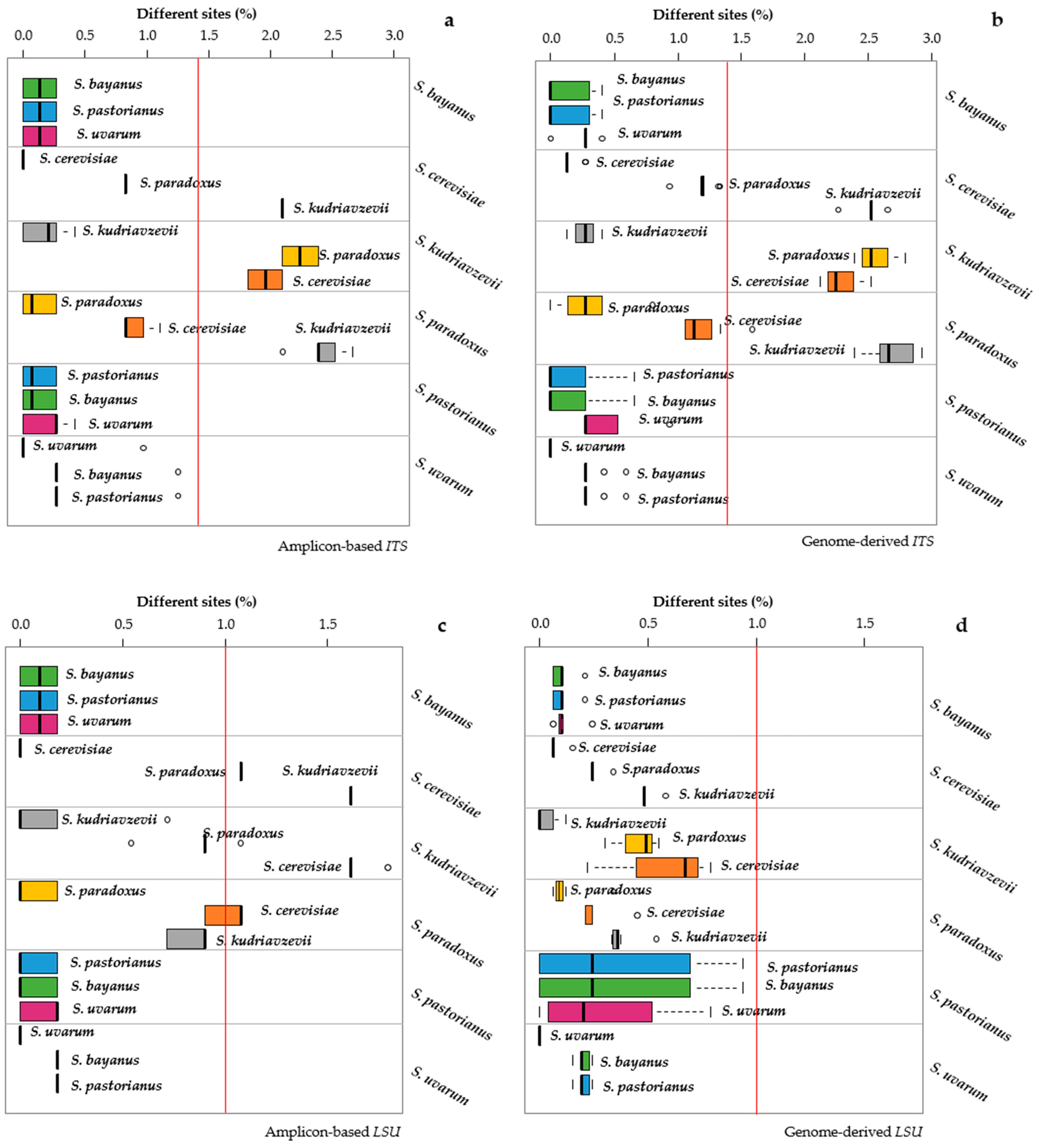{kind=link}
{kind=link}
{kind=link}
{kind=link}
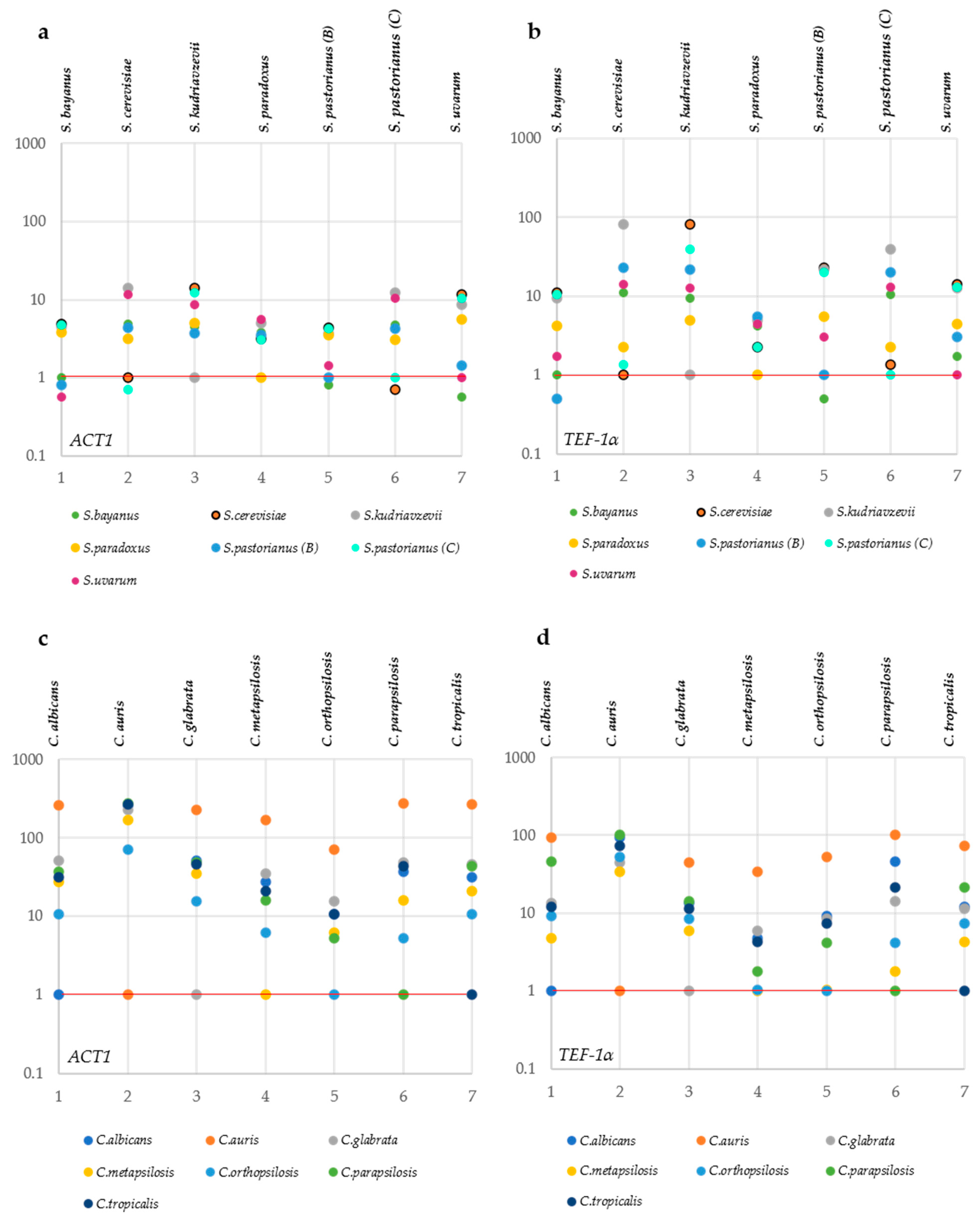{kind=link}
{kind=link}
| S. bayanus | S. cerevisiae | S. kudriavzevii | |||
| GCA_001298625.1 | GCA_003086655.1 | GCA_000167075.2 | |||
| GCA_001515405.2 | GCA_004328465.1 | GCA_000256825.1 | |||
| GCA_003327605.1 | GCA_000662435.2 | GCA_000257025.1 | |||
| GCA_013180675.1 | GCA_000976845.3 | GCA_000256985.1 | |||
| GCA_013180065.1 | GCA_000977385.2 | GCA_900682665.1 | |||
| GCA_013180125.1 | GCA_000977715.4 | GCA_000257045.1 | |||
| GCA_013180165.1 | GCA_003275125.1 | GCA_000256845.1 | |||
| GCA_013180695.1 | GCA_002571405.2 | GCA_000257085.1 | |||
| GCA_003274825.1 | GCA_000257105.1 | ||||
| GCA_009738405.1 | GCA_003327635.1 | ||||
| S. pastorianus | S. uvarum | S. paradoxus | |||
| GCA_001515445.2 | GCA_000167035.1 | GCA_002079055.1 | |||
| GCA_011022315.1 | GCA_013265775.1 | GCA_004353035.1 | |||
| GCA_013180355.1 | GCA_013179955.1 | GCA_004353095.1 | |||
| GCA_013180735.1 | GCA_013180055.1 | GCA_004353105.1 | |||
| GCA_013179865.1 | GCA_013180345.1 | GCA_000166955.1 | |||
| GCA_000805465.1 | GCA_013179815.1 | GCA_004352945.1 | |||
| GCA_001515425.2 | GCA_013265705.1 | GCA_004352955.1 | |||
| GCA_001483335.1 | GCA_013180195.1 | GCA_004352965.1 | |||
| GCA_001640265.1 | GCA_013180235.1 | GCA_009805645.1 | |||
| GCA_003004515.1 | GCA_013179965.1 | GCA_002079145.1 | |||
| C. albicans | C. auris | C. glabrata | C. metapsilosis | ||
| GCA_000182965.3 | GCA_003013715.2. | GCA_000002545.2 | GCA_008904905.1 | ||
| GCA_002837675.1 | GCA_008275145.1 | GCA_002219185.1 | GCA_900069165.1 | ||
| GCA_003454735.1 | GCA_014217455.1 | GCA_002219195.1 | |||
| GCA_005890765.1 | GCA_014673535.1 | GCA_010111755.1 | |||
| C. orthopsilosis | C. parapsilosis | C. tropicalis | |||
| GCA_000304155.1 | GCA_000982555.2 | GCA_000633855.1 | |||
| GCA_000315875.1 | GCA_011316035.2 | GCA_002864075.1 | |||
| GCA_004334915.1 | GCA_014049445.1 | GCA_006942135.1 | |||
| GCA_900002835.2 | GCA_014049495.1 | GCA_013177555.1 | |||
| Species | Marker Sequences | ||
|---|---|---|---|
| ACT1 | ITS | LSU | |
| C. albicans | AJ389057 | AB032172 | U45776 |
| C. auris | AJ389073 | AB375772 | AB375773 |
| C. glabrata | AJ389073 | AY046165 | U44808 |
| C. metapsilosis | AJ508485 | FJ872019 | AY497667 |
| C. parapsilosis | AJ508485 | KP054272 | U45754 |
| C. orthopsilosis | AJ508485 | FJ872018 | FJ746056 |
| C. tropicalis | AJ508499 | AF287910 | U45749 |
| S. cerevisiae | AJ389075 | AY046146 | AY048154 |
| RPB1 | RPB2 | TEF1-α | |
| C. albicans | JQ713048 | XM_713079.2 | AF402066 |
| C. auris | MK294611.1 | XM_029033121.1 | AF402029 |
| C. glabrata | AY497705 | AF527898 | AF402029 |
| C. metapsilosis | LN680790.1:15517-16901 | LN680773.1: 56482-58821 | LN680790.1:1502560-1503683 |
| C. parapsilosis | XM_714321.2 | JQ698980 | AF402066 |
| C. orthopsilosis | LN680790.1:15517-16901 | LN680773.1: 56482-58821 | LN680790.1:1502560-1503683 |
| C. tropicalis | CP017630.1:2260358-2265544 | CP017623.1:319665-323369 | AF402066 |
| S. cerevisiae | JQ713023 | JQ698955 | AF402004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, A.; Corte, L.; Casagrande Pierantoni, D.; Robert, V.; Cardinali, G. What Is the Best Lens? Comparing the Resolution Power of Genome-Derived Markers and Standard Barcodes. Microorganisms 2021, 9, 299. https://doi.org/10.3390/microorganisms9020299
Conti A, Corte L, Casagrande Pierantoni D, Robert V, Cardinali G. What Is the Best Lens? Comparing the Resolution Power of Genome-Derived Markers and Standard Barcodes. Microorganisms. 2021; 9(2):299. https://doi.org/10.3390/microorganisms9020299
Chicago/Turabian StyleConti, Angela, Laura Corte, Debora Casagrande Pierantoni, Vincent Robert, and Gianluigi Cardinali. 2021. "What Is the Best Lens? Comparing the Resolution Power of Genome-Derived Markers and Standard Barcodes" Microorganisms 9, no. 2: 299. https://doi.org/10.3390/microorganisms9020299
APA StyleConti, A., Corte, L., Casagrande Pierantoni, D., Robert, V., & Cardinali, G. (2021). What Is the Best Lens? Comparing the Resolution Power of Genome-Derived Markers and Standard Barcodes. Microorganisms, 9(2), 299. https://doi.org/10.3390/microorganisms9020299