
Immune-Based Anti-Staphylococcal Therapeutic Approaches


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune Boosting Strategies
2.1. Targeting Neutrophils and Related Pathways
2.1.1. Neutrophil Store
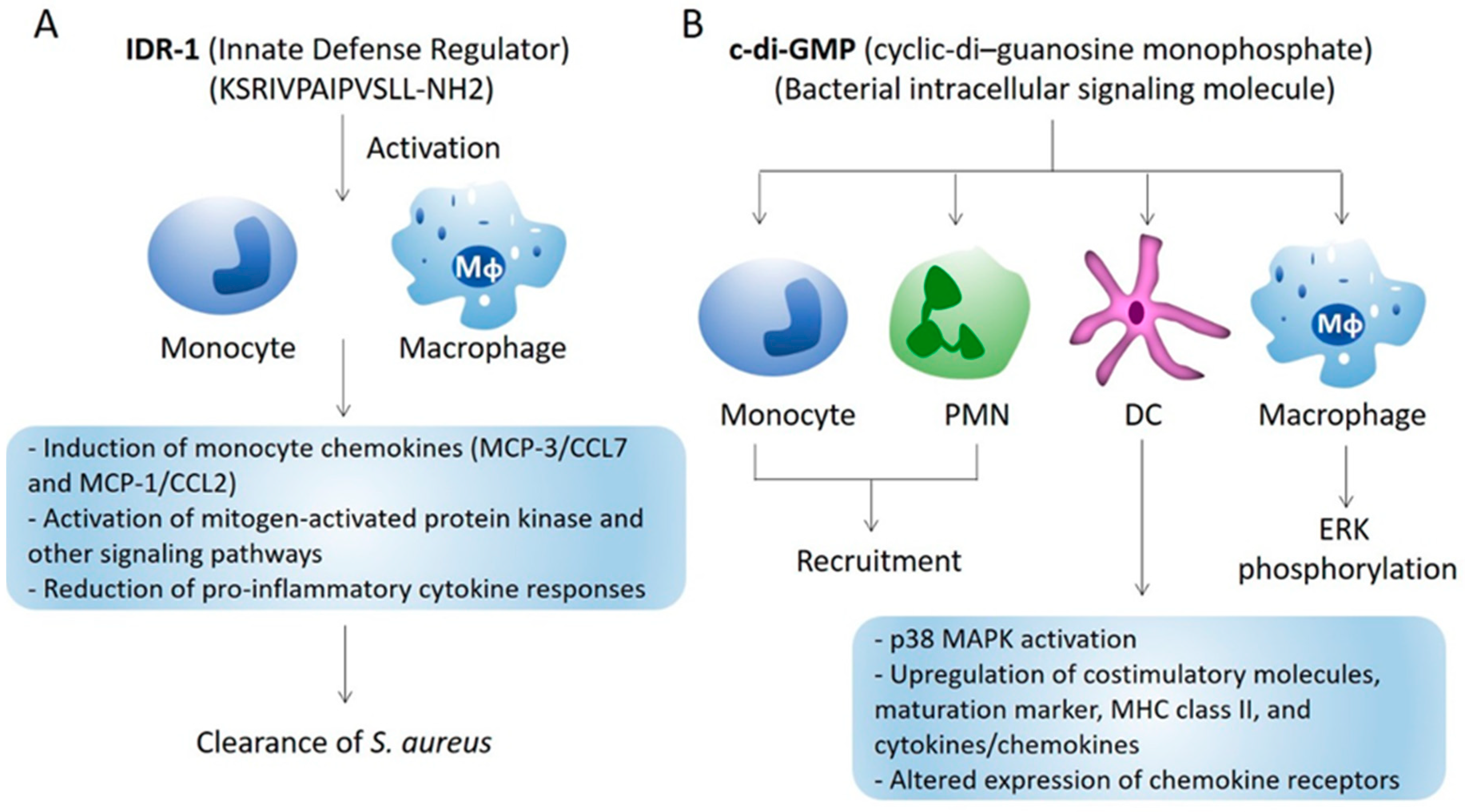
2.1.2. Myeloid Cell Recruitment
2.2. Targeting Specific Neutrophil Functions
2.2.1. ROS and RNS
2.2.2. Antimicrobial Peptides
2.2.3. NETS
2.3. Targeting Regulators of Neutrophils
2.4. Beyond Neutrophils
2.4.1. Cell Transfusion with Platelets, Macrophages, MSCs
2.4.2. Training and Reprograming the Innate Immune System
2.5. Targeting Microbial Components with Antibody Therapeutics
2.5.1. Serum Therapy
2.5.2. Enhancing Opsonophagocytosis
2.5.3. Neutralization of Toxins
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathers, C.; Stevens, G.; Hogan, D.; Mahanani, W.R.; Ho, J. Global and Regional Causes of Death: Patterns and Trends, 2000–2015. In Disease Control Priorities: Improving Health and Reducing Poverty; Jamison, D.T., Gelband, H., Horton, S., Jha, P., Laxminarayan, R., Mock, C.N., Nugent, R., Eds.; World Bank: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Nathan, C. Antibiotics at the crossroads. Nature 2004, 431, 899–902. [Google Scholar] [CrossRef]
- Thomsen, I.P.; Liu, G.Y. Targeting fundamental pathways to disrupt Staphylococcus aureus survival: Clinical implications of recent discoveries. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Naimi, T.S.; LeDell, K.H.; Como-Sabetti, K.; Borchardt, S.M.; Boxrud, D.J.; Etienne, J.; Johnson, S.K.; Vandenesch, F.; Fridkin, S.; O’Boyle, C.; et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003, 290, 2976–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, H.F. Community-associated MRSA--resistance and virulence converge. N. Engl. J. Med. 2005, 352, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Deresinski, S. Vancomycin: Does it still have a role as an antistaphylococcal agent? Expert Rev. Anti Infect. Ther. 2007, 5, 393–401. [Google Scholar] [CrossRef]
- Gould, I.M. Clinical relevance of increasing glycopeptide MICs against Staphylococcus aureus. Int. J. Antimicrob. Agents 2008, 31 (Suppl. S2), 1–9. [Google Scholar] [CrossRef]
- Steinkraus, G.; White, R.; Friedrich, L. Vancomycin MIC creep in non-vancomycin-intermediate Staphylococcus aureus (VISA), vancomycin-susceptible clinical methicillin-resistant S. aureus (MRSA) blood isolates from 2001–2005. J. Antimicrob. Chemother. 2007, 60, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deresinski, S. Counterpoint: Vancomycin and Staphylococcus aureus—An antibiotic enters obsolescence. Clin. Infect. Dis. 2007, 44, 1543–1548. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Gould, I.M.; Bal, A.M. New antibiotic agents in the pipeline and how they can help overcome microbial resistance. Virulence 2013, 4, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.; Iwase, T.; Liu, G.Y. Intranasal application of S. epidermidis prevents colonization by methicillin-resistant Staphylococcus aureus in mice. PLoS ONE 2011, 6, e25880. [Google Scholar] [CrossRef] [Green Version]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [Google Scholar] [CrossRef]
- Gordon, R.J.; Lowy, F.D. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008, 46 (Suppl. S5), S350–S359. [Google Scholar] [CrossRef] [Green Version]
- Committee on New Directions in the Study of Antimicrobial Therapeutics: New Classes of Antimicrobials, Committee on New Directions in the Study of Antimicrobial Therapeutics: Immunomodulation. In Treating Infectious Diseases in a Microbial World: Report of Two Workshops on Novel Antimicrobial Therapeutics; National Academies Press: Washington, DC, USA, 2006; pp. 21–22. [CrossRef]
- Gombart, A.F.; Koeffler, H.P. Neutrophil specific granule deficiency and mutations in the gene encoding transcription factor C/EBP(epsilon). Curr. Opin. Hematol. 2002, 9, 36–42. [Google Scholar] [CrossRef]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.H. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol. Rev. 2017, 41, 139–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlius, J.; Herbst, C.; Reiser, M.; Schwarzer, G.; Engert, A. Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst. Rev. 2008, CD003189. [Google Scholar] [CrossRef]
- Freifeld, A.G.; Bow, E.J.; Sepkowitz, K.A.; Boeckh, M.J.; Ito, J.I.; Mullen, C.A.; Raad, I.I.; Rolston, K.V.; Young, J.A.; Wingard, J.R.; et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the infectious diseases society of america. Clin. Infect. Dis. 2011, 52, e56–e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, M.G.; Peters, C.; Wacker, A.; Northoff, H.; Moog, R.; Boehme, A.; Silling, G.; Grimminger, W.; Einsele, H. Randomized phase III study of granulocyte transfusions in neutropenic patients. Bone Marrow Transplant. 2008, 42, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estcourt, L.J.; Stanworth, S.J.; Hopewell, S.; Doree, C.; Trivella, M.; Massey, E. Granulocyte transfusions for treating infections in people with neutropenia or neutrophil dysfunction. Cochrane Database Syst. Rev. 2016, 4, CD005339. [Google Scholar] [CrossRef]
- Spellberg, B.J.; Collins, M.; French, S.W.; Edwards, J.E., Jr.; Fu, Y.; Ibrahim, A.S. A phagocytic cell line markedly improves survival of infected neutropenic mice. J. Leukoc. Biol. 2005, 78, 338–344. [Google Scholar] [CrossRef]
- Panaro, M.A.; Mitolo, V. Cellular responses to FMLP challenging: A mini-review. Immunopharmacol. Immunotoxicol. 1999, 21, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Postma, B.; Poppelier, M.J.; van Galen, J.C.; Prossnitz, E.R.; van Strijp, J.A.; de Haas, C.J.; van Kessel, K.P. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J. Immunol. 2004, 172, 6994–7001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggar, A.; Ehrnfelt, C.; Holgersson, J.; Flock, J.I. The extracellular adherence protein from Staphylococcus aureus inhibits neutrophil binding to endothelial cells. Infect. Immun. 2004, 72, 6164–6167. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.G.; Dullaghan, E.; Mookherjee, N.; Glavas, N.; Waldbrook, M.; Thompson, A.; Wang, A.; Lee, K.; Doria, S.; Hamill, P.; et al. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 2007, 25, 465–472. [Google Scholar] [CrossRef]
- Lee, H.Y.; Bae, Y.S. The anti-infective peptide, innate defense-regulator peptide, stimulates neutrophil chemotaxis via a formyl peptide receptor. Biochem. Biophys. Res. Commun. 2008, 369, 573–578. [Google Scholar] [CrossRef]
- McWhirter, S.M.; Barbalat, R.; Monroe, K.M.; Fontana, M.F.; Hyodo, M.; Joncker, N.T.; Ishii, K.J.; Akira, S.; Colonna, M.; Chen, Z.J.; et al. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med. 2009, 206, 1899–1911. [Google Scholar] [CrossRef]
- Karaolis, D.K.; Means, T.K.; Yang, D.; Takahashi, M.; Yoshimura, T.; Muraille, E.; Philpott, D.; Schroeder, J.T.; Hyodo, M.; Hayakawa, Y.; et al. Bacterial c-di-GMP is an immunostimulatory molecule. J. Immunol. 2007, 178, 2171–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.M. Chronic granulomatous disease. Clin. Rev. Allergy Immunol. 2010, 38, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Ezekowitz, R.A.; Orkin, S.H.; Newburger, P.E. Recombinant interferon gamma augments phagocyte superoxide production and X-chronic granulomatous disease gene expression in X-linked variant chronic granulomatous disease. J. Clin. Investig. 1987, 80, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Ahlin, A.; Larfars, G.; Elinder, G.; Palmblad, J.; Gyllenhammar, H. Gamma interferon treatment of patients with chronic granulomatous disease is associated with augmented production of nitric oxide by polymorphonuclear neutrophils. Clin. Diagn. Lab. Immunol. 1999, 6, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Hancock, R.E. Cationic host defence peptides: Innate immune regulatory peptides as a novel approach for treating infections. Cell Mol. Life Sci. 2007, 64, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Schauber, J.; Dorschner, R.A.; Yamasaki, K.; Brouha, B.; Gallo, R.L. Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology 2006, 118, 509–519. [Google Scholar] [CrossRef]
- Buchau, A.S.; Schauber, J.; Hultsch, T.; Stuetz, A.; Gallo, R.L. Pimecrolimus enhances TLR2/6-induced expression of antimicrobial peptides in keratinocytes. J. Investig. Dermatol. 2008, 128, 2646–2654. [Google Scholar] [CrossRef] [Green Version]
- Schauber, J.; Dorschner, R.A.; Coda, A.B.; Buchau, A.S.; Liu, P.T.; Kiken, D.; Helfrich, Y.R.; Kang, S.; Elalieh, H.Z.; Steinmeyer, A.; et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J. Clin. Investig. 2007, 117, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Matheson, E.M.; Mainous, A.G., 3rd; Hueston, W.J.; Diaz, V.A.; Everett, C.J. Vitamin D and methicillin-resistant Staphylococcus aureus nasal carriage. Scand. J. Infect. Dis. 2010, 42, 455–460. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Chow, O.A.; von Kockritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Zinkernagel, A.S.; Peyssonnaux, C.; Johnson, R.S.; Nizet, V. Pharmacologic augmentation of hypoxia-inducible factor-1alpha with mimosine boosts the bactericidal capacity of phagocytes. J. Infect. Dis. 2008, 197, 214–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyme, P.; Thoennissen, N.H.; Tseng, C.W.; Thoennissen, G.B.; Wolf, A.J.; Shimada, K.; Krug, U.O.; Lee, K.; Muller-Tidow, C.; Berdel, W.E.; et al. C/EBPepsilon mediates nicotinamide-enhanced clearance of Staphylococcus aureus in mice. J. Clin. Investig. 2012, 122, 3316–3329. [Google Scholar] [CrossRef] [Green Version]
- Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J. Mol. Med. 2007, 85, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, R.; Barlow, C.; Lekstrom-Himes, J.; Castilla, L.H.; Liu, P.P.; Eckhaus, M.; Decker, T.; Wynshaw-Boris, A.; Xanthopoulos, K.G. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13187–13192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gombart, A.F.; Kwok, S.H.; Anderson, K.L.; Yamaguchi, Y.; Torbett, B.E.; Koeffler, H.P. Regulation of neutrophil and eosinophil secondary granule gene expression by transcription factors C/EBP epsilon and PU.1. Blood 2003, 101, 3265–3273. [Google Scholar] [CrossRef] [Green Version]
- LeClaire, R.D.; Kell, W.; Bavari, S.; Smith, T.J.; Hunt, R.E. Protective effects of niacinamide in staphylococcal enterotoxin-B-induced toxicity. Toxicology 1996, 107, 69–81. [Google Scholar] [CrossRef]
- Tacke, R.; Sun, J.; Uchiyama, S.; Polovina, A.; Nguyen, D.G.; Nizet, V. Protection Against Lethal Multidrug-Resistant Bacterial Infections Using Macrophage Cell Therapy. Infect. Microbes Dis. 2019, 1, 61–69. [Google Scholar] [CrossRef]
- Nemeth, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Yeaman, M.R. Platelets: At the nexus of antimicrobial defence. Nat. Rev. Microbiol. 2014, 12, 426–437. [Google Scholar] [CrossRef]
- Farghali, H.A.; AbdElKader, N.A.; AbuBakr, H.O.; Aljuaydi, S.H.; Khattab, M.S.; Elhelw, R.; Elhariri, M. Antimicrobial action of autologous platelet-rich plasma on MRSA-infected skin wounds in dogs. Sci. Rep. 2019, 9, 12722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.Y.; Yin, J.M.; Ding, H.; Jia, W.T.; Zhang, C.Q. Efficacy of leukocyte- and platelet-rich plasma gel (L-PRP gel) in treating osteomyelitis in a rabbit model. J. Orthop. Res. 2013, 31, 949–956. [Google Scholar] [CrossRef]
- Waniczek, D.; Kozowicz, A.; Muc-Wierzgon, M.; Kokot, T.; Swietochowska, E.; Nowakowska-Zajdel, E. Adjunct methods of the standard diabetic foot ulceration therapy. Evid. Based Complement. Alternat. Med. 2013, 2013, 243568. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, F.; Hu, X.; Lu, J.; Sun, X.; Gao, J.; Ling, D. Responsive Assembly of Silver Nanoclusters with a Biofilm Locally Amplified Bactericidal Effect to Enhance Treatments against Multi-Drug-Resistant Bacterial Infections. ACS Cent. Sci. 2019, 5, 1366–1376. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Chen, A.F.; Hirsch, D.; Rothenberg, A.C.; Tan, J.; Alexander, P.G.; Tuan, R.S. Antimicrobial activity of mesenchymal stem cells against Staphylococcus aureus. Stem Cell Res. Ther. 2020, 11, 293. [Google Scholar] [CrossRef]
- Yuan, Y.; Lin, S.; Guo, N.; Zhao, C.; Shen, S.; Bu, X.; Ye, H. Marrow mesenchymal stromal cells reduce methicillin-resistant Staphylococcus aureus infection in rat models. Cytotherapy 2014, 16, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Peralta, O.A.; Carrasco, C.; Vieytes, C.; Tamayo, M.J.; Munoz, I.; Sepulveda, S.; Tadich, T.; Duchens, M.; Melendez, P.; Mella, A.; et al. Safety and efficacy of a mesenchymal stem cell intramammary therapy in dairy cows with experimentally induced Staphylococcus aureus clinical mastitis. Sci. Rep. 2020, 10, 2843. [Google Scholar] [CrossRef] [Green Version]
- Mulder, W.J.M.; Ochando, J.; Joosten, L.A.B.; Fayad, Z.A.; Netea, M.G. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 2019, 18, 553–566. [Google Scholar] [CrossRef]
- Ciarlo, E.; Heinonen, T.; Theroude, C.; Asgari, F.; Le Roy, D.; Netea, M.G.; Roger, T. Trained Immunity Confers Broad-Spectrum Protection Against Bacterial Infections. J. Infect. Dis. 2020, 222, 1869–1881. [Google Scholar] [CrossRef]
- Zhu, H.; Lin, J.; Wei, H.; Bao, B.; Gao, T.; Zheng, X. Does Training Innate Immunity Confer Broad-spectrum Protection Against Bone and Joint Infection in a Mouse Model? Clin. Orthop. Relat. Res. 2020, 478, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, L.R.; Joshi, G.S.; Richardson, A.R. Peroxisome Proliferator-Activated Receptor gamma Is Essential for the Resolution of Staphylococcus aureus Skin Infections. Cell Host Microbe 2018, 24, 261–270.e264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurlow, L.R.; Joshi, G.S.; Clark, J.R.; Spontak, J.S.; Neely, C.J.; Maile, R.; Richardson, A.R. Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe 2013, 13, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.S.; Fowler, V.G.; Shukla, S.K.; Rose, W.E.; Proctor, R.A. Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol. Rev. 2020, 44, 123–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, V.G., Jr.; Proctor, R.A. Where does a Staphylococcus aureus vaccine stand? Clin. Microbiol. Infect. 2014, 20 (Suppl. S5), 66–75. [Google Scholar] [CrossRef] [Green Version]
- Pier, G.B. Will there ever be a universal Staphylococcus aureus vaccine? Hum. Vaccin. Immunother. 2013, 9, 1865–1876. [Google Scholar] [CrossRef] [Green Version]
- Casadevall, A.; Scharff, M.D. Return to the past: The case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 1995, 21, 150–161. [Google Scholar] [CrossRef]
- Francois, B.; Barraud, O.; Jafri, H.S. Antibody-based therapy to combat Staphylococcus aureus infections. Clin. Microbiol. Infect. 2017, 23, 219–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obonyo, C.O.; Lau, J. Efficacy of Haemophilus influenzae type b vaccination of children: A meta-analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 90–97. [Google Scholar] [CrossRef]
- Berman-Rosa, M.; O’Donnell, S.; Barker, M.; Quach, C. Efficacy and Effectiveness of the PCV-10 and PCV-13 Vaccines against Invasive Pneumococcal Disease. Pediatrics 2020, 145. [Google Scholar] [CrossRef]
- Fattom, A.I.; Horwith, G.; Fuller, S.; Propst, M.; Naso, R. Development of StaphVAX, A polysaccharide conjugate vaccine against S. aureus infection: From the lab bench to phase III clinical trials. Vaccine 2004, 22, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Fattom, A.; Matalon, A.; Buerkert, J.; Taylor, K.; Damaso, S.; Boutriau, D. Efficacy profile of a bivalent Staphylococcus aureus glycoconjugated vaccine in adults on hemodialysis: Phase III randomized study. Hum. Vaccin. Immunother. 2015, 11, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Stranger-Jones, Y.K.; Bae, T.; Schneewind, O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2006, 103, 16942–16947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.S.; Miller, A.A.; Donald, R.G.; Scully, I.L.; Nanra, J.S.; Cooper, D.; Jansen, K.U. Development of a multicomponent Staphylococcus aureus vaccine designed to counter multiple bacterial virulence factors. Hum. Vaccin. Immunother. 2012, 8, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.K.; Schelonka, R.; White, R.; Holley, H.P.; Bifano, E.; Cummings, J.; Adcock, K.; Kaufman, D.; Puppala, B.; Riedel, P.; et al. A blinded, randomized, multicenter study of an intravenous Staphylococcus aureus immune globulin. J. Perinatol. 2006, 26, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Rupp, M.E.; Holley, H.P., Jr.; Lutz, J.; Dicpinigaitis, P.V.; Woods, C.W.; Levine, D.P.; Veney, N.; Fowler, V.G., Jr. Phase II, randomized, multicenter, double-blind, placebo-controlled trial of a polyclonal anti-Staphylococcus aureus capsular polysaccharide immune globulin in treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2007, 51, 4249–4254. [Google Scholar] [CrossRef] [Green Version]
- Bergdoll, M.S.; Crass, B.A.; Reiser, R.F.; Robbins, R.N.; Lee, A.C.; Chesney, P.J.; Davis, J.P.; Vergeront, J.M.; Wand, P.J. An enterotoxin-like protein in Staphylococcus aureus strains from patients with toxic shock syndrome. Ann. Intern. Med. 1982, 96, 969–971. [Google Scholar] [CrossRef]
- Parsonnet, J.; Hansmann, M.A.; Delaney, M.L.; Modern, P.A.; Dubois, A.M.; Wieland-Alter, W.; Wissemann, K.W.; Wild, J.E.; Jones, M.B.; Seymour, J.L.; et al. Prevalence of toxic shock syndrome toxin 1-producing Staphylococcus aureus and the presence of antibodies to this superantigen in menstruating women. J. Clin. Microbiol. 2005, 43, 4628–4634. [Google Scholar] [CrossRef] [Green Version]
- Fritz, S.A.; Tiemann, K.M.; Hogan, P.G.; Epplin, E.K.; Rodriguez, M.; Al-Zubeidi, D.N.; Bubeck Wardenburg, J.; Hunstad, D.A. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin. Infect. Dis. 2013, 56, 1554–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus alpha-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [Green Version]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubeck Wardenburg, J.; Schneewind, O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 2008, 205, 287–294. [Google Scholar] [CrossRef]
- Yu, X.Q.; Robbie, G.J.; Wu, Y.; Esser, M.T.; Jensen, K.; Schwartz, H.I.; Bellamy, T.; Hernandez-Illas, M.; Jafri, H.S. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, Extended-Half-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Magyarics, Z.; Leslie, F.; Bartko, J.; Rouha, H.; Luperchio, S.; Schorgenhofer, C.; Schwameis, M.; Derhaschnig, U.; Lagler, H.; Stiebellehner, L.; et al. Randomized, Double-Blind, Placebo-Controlled, Single-Ascending-Dose Study of the Penetration of a Monoclonal Antibody Combination (ASN100) Targeting Staphylococcus aureus Cytotoxins in the Lung Epithelial Lining Fluid of Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Paterson, M.J.; Caldera, J.R.; Nguyen, C.; Sharma, P.; Castro, A.M.; Kolar, S.L.; Tsai, C.M.; Limon, J.J.; Becker, C.A.; Martins, G.A.; et al. Harnessing antifungal immunity in pursuit of a Staphylococcus aureus vaccine strategy. PLoS Pathog. 2020, 16, e1008733. [Google Scholar] [CrossRef]
- Armentrout, E.I.; Liu, G.Y.; Martins, G.A. T Cell Immunity and the Quest for Protective Vaccines against Staphylococcus aureus Infection. Microorganisms 2020, 8, 1936. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, S.E.; Sakoulas, G.; Perencevich, E.N.; Schwaber, M.J.; Karchmer, A.W.; Carmeli, Y. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: A meta-analysis. Clin. Infect. Dis. 2003, 36, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: Executive summary. Clin. Infect. Dis. 2011, 52, 285–292. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, B.; Liu, G.Y. Immune-Based Anti-Staphylococcal Therapeutic Approaches. Microorganisms 2021, 9, 328. https://doi.org/10.3390/microorganisms9020328
Park B, Liu GY. Immune-Based Anti-Staphylococcal Therapeutic Approaches. Microorganisms. 2021; 9(2):328. https://doi.org/10.3390/microorganisms9020328
Chicago/Turabian StylePark, Bonggoo, and George Y. Liu. 2021. "Immune-Based Anti-Staphylococcal Therapeutic Approaches" Microorganisms 9, no. 2: 328. https://doi.org/10.3390/microorganisms9020328