1. Introduction
Salmonella enterica comprises over 2500 serotypes that are able to infect a wide range of animal hosts causing a variety of diseases ranging from gastroenteritis to systemic infections [
1,
2]. During the course of infection,
S. enterica injects effector proteins into the cytoplasm of host cells using type III secretion systems (T3SS). This process is relevant for
Salmonella virulence, as most T3SS effectors subvert host cellular functions through their enzymatic activities and physical interactions, promoting bacterial survival and colonization (reviewed in [
2]).
S. enterica harbors two independent T3SS encoded in pathogenicity islands SPI-1 and SPI-2 (T3SS-1 and T3SS-2, respectively). At least 7 effectors are known to be secreted through T3SSI-1, 22 through T3SS-2, and 9 through both systems (reviewed in [
3]). Effectors include a signal sequence at the N-terminal region (first 20–30 residues) required for secretion through T3SS. These sequences lack a discernible consensus, which hinders the identification of possible effector proteins [
4].
The analysis of translational fusions has proven to be very useful to evaluate effector translocation into eukaryotic host cells. Sory and coworkers described a technique using the catalytic adenylate cyclase domain of the bifunctional CyaA toxin from
Bordetella pertussis (
CyaA’) [
5].
CyaA’ is a calmodulin-dependent adenylate cyclase that catalyzes conversion of ATP into cyclic AMP (cAMP). Since calmodulin is ubiquitous in eukaryotic cells but absent in bacteria, the translocation of an effector fused to
CyaA’ can be evaluated by measuring the cAMP levels in infected cells.
Recently, Ramos-Morales and coworkers developed a protocol to generate site-specific
cyaA’ translational fusions in the chromosome of
S. enterica [
6] based on the Red recombination system from bacteriophage λ [
7]. Although useful, this method presents an important limitation: because of the structure of the mutant allele encoding each fusion, undesirable polar effects may arise from antibiotic resistance gene expression. This is particularly relevant when genes encoding effector proteins are located in operons or nearby genes or operons encoding structural components of the associated T3SS. In this work, we describe a method that allows the generation of unmarked
cyaA’ translational fusions in the bacterial chromosome using the λ Red recombination system. To this end, we constructed template plasmids pCyaA’-Kan and pCyaA’-Cam that are used to amplify the PCR products required for recombination. As a proof of concept, we generated unmarked
cyaA’ translational fusion to genes encoding several T3SS effectors in the chromosome of
S. enterica serovar Typhimurium (
S. Typhimurium). The production of each fusion was evaluated by Western blot using an anti-CyaA’ monoclonal antibody, and occurred only in response to growth conditions that induce the expression of the corresponding native gene. In addition, T3SS-1-dependent secretion of a SipA-CyaA’ fusion during in vitro growth was evidenced by Western blot analysis of culture supernatants. Finally, translocation of the SipA-CyaA’ fusion into HeLa cells was confirmed by measuring cAMP levels in infected cells.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
The bacterial strains used in this study are listed in
Table S1. All
S. Typhimurium strains are derivatives of the wild-type, virulent strain 14028s [
8,
9]. Bacteria were routinely grown in Luria-Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) at 37 °C with agitation (180 rpm). If bacteria harbored a temperature-sensitive plasmid (i.e., pKD46 or pCP20), incubations were performed at 30 °C. When required, media were supplemented with ampicillin (Amp, 100 mg/L), kanamycin (Kan, 75 mg/L), or chloramphenicol (Cam, 20 mg/L). Media were solidified by the addition of agar (15 g/L). For SPI-1-inducing conditions, bacteria were grown at 37 °C without agitation in LB medium containing 300 mM NaCl. For SPI-2-inducing conditions, bacteria were grown at 37 °C with agitation (180 rpm) in N-minimal medium (5 mM KCl, 0.5 mM (NH
4)
2SO
4, 0.5 mM K
2SO
4, 1 mM KH
2PO
4, 10 μM MgCl
2) [
10] buffered in 100 mM MES (pH 5.8) and supplemented with 0.1% casamino acids and 0.4% glucose as carbon source.
2.2. Standard DNA Techniques
Plasmid DNA was obtained using the QIAprep Spin Miniprep kit (Qiagen, Germantown, MD, USA). PCR products were purified using the QIAquick PCR Purification kit (Qiagen, Germantown, MD, USA). DNA digestions using restriction endonucleases
BamHI,
XhoI and
SpeI (New England BioLabs, Ipswich, MA, USA) and ligations using T4 DNA ligase (New England BioLabs, Ipswich, MA, USA) were conducted as recommended by the manufacturer. When required, DNA fragments from digestions were purified from 1% agarose gels prepared in Tris-acetate-EDTA (TAE) buffer using the QIAquick Gel Extraction kit (Qiagen, Germantown, MD, USA). DNA samples were routinely analyzed by electrophoresis in 1% agarose gels prepared in TAE buffer and visualized under UV light after GelRed (Biotium Inc., Fremont, CA, USA) staining. Primers used in this study are listed in
Table S2.
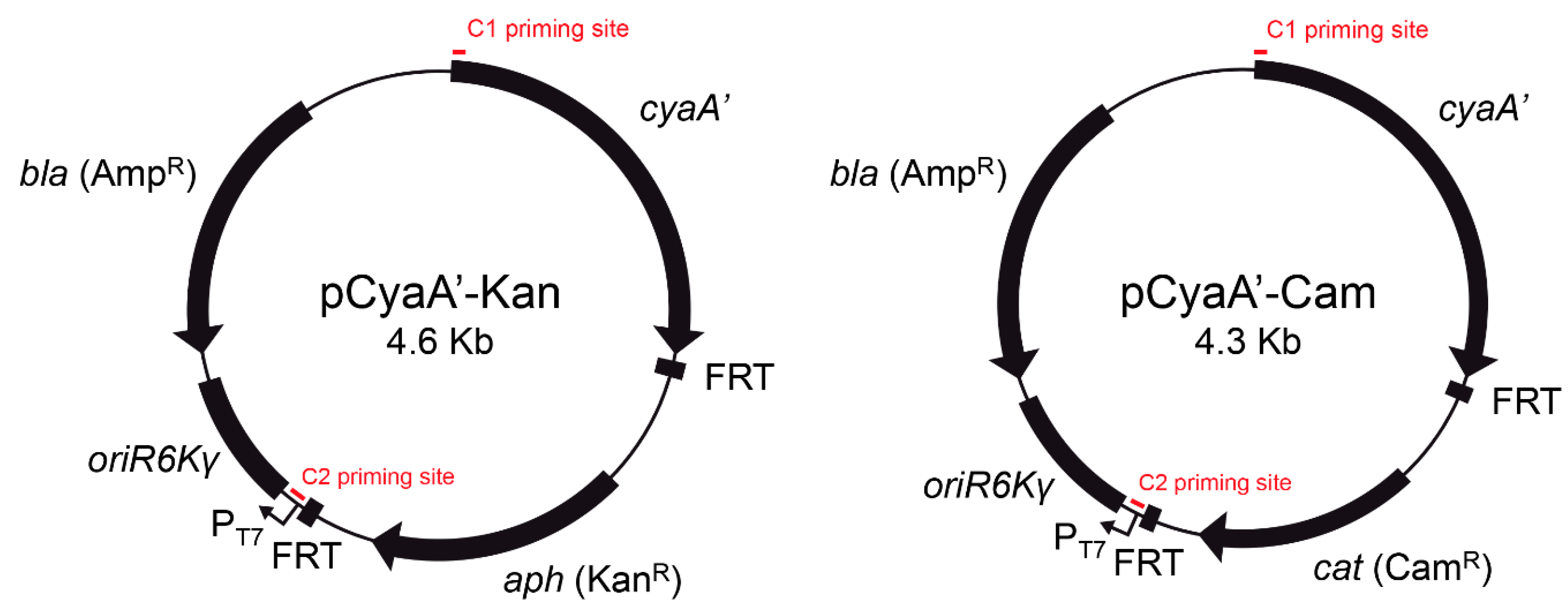
2.3. Construction of Plasmids pCyaA’-Kan and pCyaA’-Cam
To generate pCyaA’-Kan, the
cyaA’ region was amplified from pUTmini-Tn
5cyaA’ [
6] using primers cyaA(F)-BamHI and cyaA(R)-XhoI (
Table S2), and the PCR product was cloned into pGEM-T Easy (Promega, Madison, WI, USA) as recommended by the manufacturer to generate pGEM-T::
cyaA’. The Kan resistance cassette flanked by Flp recombinase target (FRT) sites was amplified from pCLF4 (GenBank EU629214) [
11] using primers pCLF4(F)-XhoI and pCLF4(R)-BamHI-XhoI (
Table S2), and the PCR product was cloned into pGEM-T Easy as recommended by the manufacturer to generate pGEM-T::Kan. A DNA fragment containing the Kan resistance cassette flanked by FRT sites was obtained by digestion of pGEM-T::Kan with
XhoI, and cloned into the unique
XhoI site downstream of
cyaA’ in pGEM-T::
cyaA’ to generate pGEM-T::
cyaA’-Kan. The orientation of the insert in the resulting plasmid was checked by PCR using primers cyaA(F)-BamHI and pCLF4(R)-BamHI-XhoI (
Table S2). A DNA fragment containing
oriR6K and
bla gene was amplified from pKD4 (GenBank A Y048743) [
7] using primers pCLF4(F)-BamHI and pCLF4(R)-BamHI (
Table S2). The PCR product was purified and digested with
BamHI. Finally, this fragment was ligated to a fragment containing
cyaA’ and the Kan resistance cassette flanked by FRT sites obtained by digestion of pGEM-T::
cyaA’-Kan with
BamHI.
To generate pCyaA’-Cam, the backbone of pCLF2 (GenBank HM047089) was amplified using primers pCLF4(F)-XhoI and pCLF4(R)-SpeI (
Table S2) to incorporate unique
XhoI and
SpeI sites. The PCR product was purified, digested with
XhoI and
SpeI, and ligated to a DNA fragment containing
cyaA’ obtained by digestion of pGEM-T::
cyaA’ with
XhoI and
SpeI.
Plasmids pCyaA’-Kan and pCyaA’-Cam carry the R6Kγ replication origin, which requires the trans-acting π protein (encoded by pir) for replication. So, they were propagated in Escherichia coli DH5α λpir. Derivatives of plasmid pGEM-T Easy were propagated in E. coli DH5α.
2.4. Generation of cyaA’ Translational Fusions
Derivatives of
S. Typhimurium 14028s containing chromosomal fusions of
cyaA’ to genes encoding effectors secreted by T3SS-1 (
sipA,
sptP, and
sopB), T3SS-2 (
sifA,
sseJ,
sopD2,
steC, and
sseG), or both T3SS-1 and T3SS-2 (
spvB and
gtgE) were constructed by the Red-swap recombination method [
7], with modifications. Briefly, a DNA fragment including the
cyaA’ ORF and a Kan resistance cassette was amplified from plasmid pCyaA’-Kan using specific primers (“xxx_H1 + C1” and “xxx_H2 + C2”) designed for each fusion (
Table S2). Alternatively, the same primers were used to amplify a DNA fragment including the
cyaA’ ORF and a Cam resistance cassette from plasmid pCyaA’-Cam.
S. Typhimurium 14028s carrying the temperature-sensitive plasmid pKD46, which expresses the λ Red recombinase system, was grown to an OD
600 of 0.5 at 30 °C in LB medium supplemented with Amp and L-arabinose (10 mM). Bacteria were made electrocompetent by sequential washes with ice-cold sterile 15% glycerol, and transformed with ~500 ng of each PCR product. Transformants were selected on LB agar supplemented with Kan or Cam at 37 °C. The presence of each chromosomal fusion was confirmed by PCR amplification using specific “forward” primers (“xxx_Out5”) designed for each effector together with “reverse” primer CyaRev that hybridizes within
cyaA’ (
Table S2).
To obtain non-polar unmarked
cyaA’ translational fusions, the corresponding antibiotic resistance cassette was removed by transforming each mutant with the temperature-sensitive plasmid pCP20, which encodes the Flp recombinase [
7,
12]. Transformants were selected on LB agar supplemented with Amp at 30 °C. Next, individual colonies were replica-plated on LB agar, LB agar supplemented with Amp, and LB agar supplemented with Kan or Cam, and incubated at 37 °C. Transformants that had lost pCP20 and the corresponding antibiotic resistance cassette were identified as those unable to grow in the presence of Amp and Kan or Cam. The absence of the antibiotic resistance cassette was confirmed by PCR amplification using primers cyaA(F)-BamHI and pCLF4(R)-BamHI-XhoI (
Table S2). Finally, phage P22 HT105-1
int-201 was used to transduce mutant alleles ∆
invA::Kan and ∆
ssaD::Kan into a derivative of
S. Typhimurium 14028s harboring an unmarked sipA-cyaA’ chromosomal fusion to inactive T3SS-1 or T3SS-2, respectively. The presence of each mutant allele was confirmed by PCR amplification using primers flanking the sites of substitution (
Table S2).
2.5. Western Blot Analyses
Different bacterial strains were grown at 37 °C for 5 h under in vitro conditions that induce the expression of SPI-1 or SPI-2 genes (10 mL cultures). For preparation of whole cell lysates, bacteria recovered from 1 mL of each culture were suspended in phosphate-buffered saline (PBS) and adjusted to an OD
600 of 2. Next, 75 μL of each bacterial suspension was mixed with 25 μL of 4 × Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) and the mix was boiled for 10 min and stored at −20 °C until further use. For preparation of secreted proteins, the supernatant from each remaining culture was passed through a 0.2-µm filter to remove residual bacteria. Proteins from the supernatants were precipitated with trichloroacetic acid (10%
v/
v) and washed 3 times with ice-cold acetone. The pellet was air dried and subjected to metanol-chloroform precipitation [
13] to remove salts, as described [
14]. The final pellet was suspended in 40 μL of 4 × Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) and the mix was boiled for 10 min and stored at −20 °C until further use.
Samples of each lysate (10 μL) or preparations of secreted proteins (20 μL) were resolved by SDS-PAGE in 12% polyacrylamide gels using a Mini-Protean III system (Bio-Rad, Hercules, CA, USA). The electrophoresis was conducted at 120 V (constant) using 1× running buffer (1.44% glycine, 0.3% Tris, 0.1% SDS). Transfer of proteins from polyacrylamide gels to polyvinylidene fluoride (PVDF) membranes was performed in a Mini Trans-Blot system (Bio-Rad, Hercules, CA, USA) for 90 min at 300 mA in transfer buffer (1.44% glycine, 0.3% Tris, 20% methanol). Membranes were incubated for 2 h at room temperature in a blocking solution containing 5% BSA (Sigma-Aldrich, St. Louis, MA, USA) in Tris-buffered saline supplemented with 0.1% Tween-20 (TBST). After blocking, the membranes were incubated overnight at 4 °C with the mouse anti-CyaA’ monoclonal antibody 3D1 (Santa Cruz Biotechnology, Dallas, TX, USA) diluted in blocking solution (1:10,000) or the mouse anti-DnaK monoclonal antibody [8E2/2] ab69617 (Abcam, Cambridge, MA, USA) diluted in blocking solution (1:10,000). After three washes with TBST, the membranes were incubated for 2 h at room temperature with an anti-mouse IgG antibody conjugated with horseradish peroxidase (Cell Signaling Technology, Danvers, MA, USA) diluted in blocking solution (1:10,000). Finally, the protein bands were revealed by using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham, MA, USA) as recommended by the manufacturer. Digital images were collected using a Dyversity 4 imaging system (Syngene, Cambridge, UK) equipped with the GeneSys 1.2.5.0 software (Syngene, Cambridge, UK).
2.6. Mammalian Cell Culture and Infection Assays
HeLa cells were routinely grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in the presence of 5% CO2. Monolayers for infection were prepared by seeding ~2 × 105 cells per well in a 24-well plate and incubating for 18 h at 37 °C in the presence of 5% CO2. Prior to infection, each monolayer was washed three times with sterile PBS. Bacteria were grown overnight at 37 °C under in vitro conditions that induce SPI-1 genes, washed three times with sterile PBS, suspended in 400 µL of DMEM-FBS, and added to monolayers of HeLa cells at a multiplicity of infection (MOI) of 100 bacteria/cell. The plate was centrifuged at 200 × g for 5 min (to facilitate the interaction of bacteria and cells) and then incubated at 37 °C in the presence of 5% CO2. After 1 h of incubation, the cells were washed two times with sterile PBS and incubated for 1 h in DMEM-FBS supplemented with gentamicin (200 µg/mL) to kill extracellular bacteria. Finally, the cells were washed three times with sterile PBS and further incubated for 1, 3, or 6 h post infection in DMEM-FBS supplemented with gentamicin (20 µg/mL).
2.7. Intracellular cAMP Measurement
Infected cell monolayers were washed three times with sterile PBS. Next, the cells were lysed using 130 µL of 1× Sample Diluent supplied with the DetectX cAMP Direct Immunoassay kit (Arbor Assays, Ann Arbor, MI, USA). Each lysate was then transferred to a microcentrifuge tube and centrifuged at 4000× g for 15 min at 4 °C. The supernatant was transferred to a clean microcentrifuge tube and stored at −20 °C until further use. Finally, the cAMP levels in each sample were determined by using the DetectX cAMP Direct Immunoassay kit (Arbor Assays, Ann Arbor, MI, USA) following the manufacturer instructions.
4. Discussion
In the present study, we describe the construction of novel template plasmids pCyaA-Kan and pCyaA-Cam (
Figure 1) for the generation of chromosomal
cyaA’ translational fusions based on site-directed integration of a PCR product using the λ Red recombination system [
7]. Unlike protocols to generate chromosomal
cyaA’ fusions described previously [
6,
15], our method allows the removal of the antibiotic resistance cassette by Flp-mediated recombination [
7,
12], resulting in an unmarked chromosomal gene fusion (
Figure 2).
Reporter gene fusions have been used as a tool to identify new effector proteins in pathogenic bacteria and for monitoring their expression, secretion, and translocation into infected cells. Several approaches involving chromosomally-encoded fusions have been developed as they are advantageous over plasmid-encoded fusions since they result in a single-copy gene fusion whose expression depends on native promoters [
6,
15,
16,
17]. In this context,
cyaA’ has arisen as an appealing reporter gene for studying the expression, secretion, and translocation of effector proteins in the case of several pathogenic bacteria [
5,
15,
18,
19,
20,
21,
22]. The advantage of using a
CyaA’ enzymatic tag is that the expression and secretion of the corresponding fusion protein can be detected in bacterial cultures in vitro by immunoblotting [
18,
21,
22], and also translocation of these fusions into host cells can be monitored by its calmodulin-dependent adenylate cyclase activity, measuring the cAMP levels in infected cells [
5,
15,
18,
19,
20,
21,
22].
Regarding
S. enterica, a strategy based on the Red recombination system from bacteriophage λ has been successfully used for the generation of site-specific
cyaA’ translational fusions in the chromosome [
6]. However, the main advantage of using our template plasmids is the possibility to generate unmarked
cyaA’ translational fusions by removal of the antibiotic resistance cassette via Flp-mediated recombination. This novel feature is highly desirable when target genes encoding effector proteins are located in operons or nearby genes encoding structural components of the associated T3SS, where undesirable polar effects may arise from strong promoter driving the expression of the antibiotic resistance cassette.
We tested the functionality of our novel template plasmids by constructing
cyaA’ fusions to 10 genes encoding T3SS effectors in the chromosome of
S. Typhimurium. In all cases, the fusion protein was detected by immunoblot in bacterial lysates from cultures grown under the corresponding expression-inducing conditions (
Figure 3 and
Figure S1). This validates the use of
CyaA’ as an epitope tag for studying expression and regulation of genes encoding T3SS effector proteins. Of note, removal of the antibiotic resistance cassette did not affect protein detection and regulated expression of the corresponding translational fusion (
Figure S2). Furthermore, using a
S. Typhimurium strain carrying an unmarked
cyaA’ fusion, we confirmed the T3SS-1-dependent secretion of SipA-CyaA’ during in vitro growth by Western blot analysis of culture supernatants (
Figure 4). We also confirmed the translocation of SipA-CyaA’ into eukaryotic cells by measuring the levels of cAMP in HeLa cells infected with the mentioned mutant strain (
Figure 5). Although not tested in this work, detection of translocated fusion proteins into eukaryotic cells could also be performed by immunoblotting after subcellular fractionation, as reported [
23], which can be an attractive alternative when using host cells with elevated phosphodiesterase activity. In addition, anti-CyaA antibodies can be used to detect the intracellular localization of a given T3SS effector fused to
CyaA’ by immunofluorescence microscopy, as described [
24,
25].
Even though this study was focused on the characterization of S. Typhimurium T3SS effectors, our procedure for generation of unmarked chromosomal cyaA’ translational fusions can be applied to any bacterial species that uses T3SS or other secretion systems to translocate effector proteins into eukaryotic cells (e.g., T4SS and T6SS), and that allows the function of λ Red and Flp recombinases (e.g., E. coli and Shigella, among others).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}