Abstract
Magnaporthe oryzae, one of the most notorious plant pathogens in the agronomic ecosystem, causes a destructive rice blast disease around the world. The blast fungus infects wide arrays of cultivated and non-cultivated plants within the Poaceae. Studies have shown that host speciation exerts selection pressure that drives the evolution and divergence of the M. oryzae population. Population genetic relationship deducted by genome-wide single nucleotide polymorphisms showed that M. oryzae differentiation is highly consistent with the host speciation process. In particular, the rice-infecting population of M. oryzae is distinct from populations from other hosts. However, how genome regions prone to host-mediated selection pressures associated with speciation in M. oryzae, especially at a large-scale population level, has not been extensively characterized. Here, we detected strong evidence of sweep selection throughout the genomes of rice and non-rice pathotypes of M. oryzae population using integrated haplotype score (iHS), cross population extended haplotype homozygosity (XPEHH), and cross population composite likelihood ratio (XPCLR) tests. Functional annotation analyses of the genes associated with host-mediated selection pressure showed that 14 pathogenicity-related genes are under positive selection pressure. Additionally, we showed that 17 candidate effector proteins are under positive and divergent selection among the blast fungus population through sweep selection analysis. Specifically, we find that a divergent selective gene, MGG_13871, is experiencing host-directed mutation in two amino acid residues in rice and non-rice infecting populations. These results provide a crucial insight into the impact of selective sweeping on the differentiation of M. oryzae populations and the dynamic influences of genomic regions in promoting host adaptation and speciation among M. oryzae species.
1. Introduction
Rice is cultivated globally and feeds more than a half of the world’s people [1]. Magnaporthe oryzae, the causal agent of rice blast disease, is a pathogenic filamentous fungus that causes a 10–30% loss of harvest each year [2,3]. M. oryzae is a multihost pathogen that can infect approximately 50 species of both wild and cultivated plants, especially cereals from the Poaceae family, such as rice, wheat, foxtail millet, and finger millet crops [4], posing a huge threat to global food security [5,6]. Since the whole genomic sequence of M. oryzae laboratory strain 70–15 was first deciphered in 2005 [7], many more isolates have been sequenced from different host plants, providing extensive resources for further research. Although the M. oryzae complex consists of genetically distinct lineages, divergence tends to be associated with host specialization [8]. In particular, the rice-infesting isolates come from a single lineage distinct from the others.
The limitation of gene flow among the different host populations is also partly responsible for M. oryzae’s host-specific subgroup divergence. For instance, it is clear that the Oryza sativa population of M. oryzae is different from others due to the lack of gene flow under asexual reproduction [9]. Nerveless, ancient recombination events and contemporary sexual reproduction might possibly occur within the Oryza isolate population [10,11]. As a seed-borne pathogen, M. oryzae can cause novel infections following seed and grain transportation both between and within countries. For instance, after wheat blast disease caused by M. oryzae isolates was first discovered in southern Brazil [12], neighboring countries such as Argentina, Bolivia, and Paraguay also detected it successively. Gene flow between different hosts has also been proposed to occur between divergent cereal- and grass-specific lineages of M.oryzae [8].
The rice blast pathogen M. oryzae engages in plant–pathogen interactions and a form of host shift. Host jumping between distant hosts, host range expansion, and host tracking between wild and domesticated hosts are sophisticated mechanisms that drive the evolutionary trend in plant pathogenic microbes [13,14,15,16]. Therefore, differentiation occurs among populations of M. oryzae from a variety of hosts that contribute to the formation of host-specific subgroups, including Oryza spp., Setaria spp., and Triticum spp. [17,18]. For instance, reports have shown that M. oryzae isolates from foxtail millet could also infect wild Setaria spp., indicating that these wild grass species serve as an alternative host that shelters the blast fungus during off seasons [19]. However, the inability of cultivated rice-infecting isolates from M. oryzae populations to infect non-cultivated wild grasses (ancestral rice species) gives credence to the postulations that M. oryzae experienced a host jump along with rice domestication approximately 10,000 years ago [4]. Domestication pressure appears to have narrowed the host range of rice-infecting M. oryzae isolates [11], which makes them distant from the Setaria-infecting isolates regarded as the closest relative of rice-infecting M. oryzae isolates [17]. Additionally, gene gain and loss resulted from the reshuffling of transposable elements throughout the individual genomes have been proposed as crucial contributors to host divergence between Magnaporthe species adapted to cultivated rice and Setaria [20]. Meanwhile, evolutionary relationship studies of M. oryzae isolates from multiple hosts have confirmed that speciation among Magnaporthe spp. coincides with the divergence of time-differing hosts [21]. To understand the evolutionary mechanisms that support the adaptation of M. oryzae to different hosts, it is crucially important to provide further insights into the sustainable management of rice blast disease. Although the mechanism underlying M. oryzae harbored by diverse hosts is still unknown, research on genomic regions that accelerate molecular evolution [22] and gene expression differentiation [9] provides an enlightening way to interpret the pathogen’s host adaptation.
Effector proteins secreted by the plant pathogen determine its pathogenicity or virulence. Therefore, there is an inevitable relationship between effector repertoire and pathogen–host specialization. It has been concluded that the specific expression patterns of effectors in three rice isolate lineages may be associated with the adaptation of M. oryzae to rice [23]. Similarly, during the interaction between barley and various host-specific lineage isolates, the effector PWT1 acts as the major host-specific gene involved in infection [24], just as the PWL1 and PWL2 genes are involved in M. oryzae’s infection of weeping lovegrass [25]. However, the secreted effector AVR1-CO39 cloned from grass isolate 2539 can be avirulence in rice with no such functional genes [26,27,28]. Directional selection induced by the various hosts also acts as a major evolutionary force that results in the host specificity of M.oryzae [29]. Comparative genomic analysis of M. oryzae and M. penniseti has suggested that the divergence of pathogenicity-related gene repertoires contributes to host adaptation [30].
Selective sweep is regarded as an essential genetic mechanism for enabling species to successfully adapt to prevailing environmental conditions by fixing novel mutations associated with beneficial phenotypic traits. This provides straightforward evidence of the evolution of populations. Diverse statistical theories and tools have been developed to identify genome-wide selection sweep events, such as F-statistics (Fst) [31], Tajima’s D-test [32], the cross-population composite likelihood ratio (XPCLR) [33], the integrated haplotype score (iHS) [34], and cross-population extended haplotype homozygosity test (XPEHH) [35]. iHS was used in a single population (within-population method) to reveal selection evidence according to the theory of linkage disequilibrium, which is effective for regions containing sites with a rapidly increased frequency of the derived allele [36]. The XPEHH and XPCLR are widely used to detect the selection footprint between two populations. Based on the difference in the multi-locus allele frequency of two populations, XPCLR shows sensitive performance regarding allele frequency fluctuations at loci with random drift [33]; in contrast, XPEHH uses linkage disequilibrium to distinguish the selection region [35]. The XPCLR, iHS, and XPEHH are widely employed to uncover genome-wide selective sweep regions. For instance, a host-driven selective sweep was identified by the XPEHH, XPCLR, and iHS tests in populations of the fungal wheat pathogen Zymoseptoria tritici from two different wheat cultivars [37]. Selective sweep throughout the genome, as detected by iHS and the XPEHH, was considered a more potent contributing force to barley scald pathogen Rhynchosporium commune through local adaptation [38]. It is likely that for the rice pathogen Xanthomonas oryzae pv. oryzae, selective sweep also had a substantial impact on the formation of the population structure [39]. The iHS, XPEHH, and XPCLR were adopted to detect the selection footprint on the X chromosome in three geographical pig breeds [40], and positive selection genes involving climate adaptation were previously identified in African cattle populations with the XPEHH and XPCLR tests [41]. The iHS, XPEHH, and XPCLR tests also revealed that positive or divergent selection genes are associated with adaptive traits in weedy rice [42]. Although genetic and environmental homogeneity in agricultural ecosystems imposes intense and uniform selection pressures on plant pathogens, the impact on the genomic variation recorded among M. oryzae populations is not fully understood.
To investigate the evolutionary mechanism that caused host adaption to emerge for M. oryzae, we collected 197 whole-genome sequences of isolates from ten different hosts, including rice, wheat, and cultivated and non-cultivated grasses. Above all, genome-wide evolution relationship analysis of M. oryzae populations revealed that host specialization crucially impacts the population structure. We showed that M. oryzae populations from rice are distinct from isolates from non-rice hosts. Furthermore, a composite strategy including the iHS, XPEHH, and XPCLR tests was adopted to identify the selective sweep within and between rice and non-rice M. oryzae populations. The selected region, spanning genes with strong positive selection signals, includes pathogenicity-related genes, such as glycosyl hydrolase, glucanase, and cutinase. Meanwhile, population-specific selective effector candidates, including MGG_15370, MGG_07993, MGG_00230, MGG_07352, MGG_06231, MGG_06234, MGG_17666, MGG_15458, MGG_05538, MGG_14374, MGG_08214, MGG_16925, MGG_15106, MGG_16938, MGG_07311, MGG_07246, and MGG_16953, were also identified in rice and non-rice populations. This study expands our understanding of host-driven population differentiation in plant–pathogen interactions.
2. Materials and Methods
2.1. Identification of Single Nucleotide Polymorphisms (SNPs)
Nucleotide sequences of 197 M. oryzae isolates previously sequenced from 10 different hosts (Supplementary Table S1) were downloaded from the National Centre for Biotechnology Information (NCBI). Subsequent comparative genomic analyses with the Mummer4 software (–maxmatch –c 100 –p) [43] identified SNPs between the 70-15 strain and the individual isolates. The SnpEff [44] tool was used to precisely extract SNPs located in the exonic regions of the 70-15 strain. VCFtools was applied to filter the variants with the following main options: –max-missing 0.50 –mac 3 –max-alleles 2 [45].
2.2. Phylogeny and Population Structure of the M. oryzae Species Complex
A neighbor-joining (NJ) tree was constructed using a Provesti’s distance [46] of 1000 bootstrap replication with the poppr package in R-3.6.1 [47]. A no-root tree with bootstrap values was visualized with the ggtree package [48]. Next, the relationships between different host-derived isolate populations were analyzed with principal component analysis (PCA) in the adegenet package [49]. The population structure was analyzed with the discriminant analysis of the principal component method using the adegenet package in R-3.6.1 [49].
2.3. Population Selective Sweep Detection
The iHS [36] and XPEHH [50] were calculated to detect the selective signature for rice and other host populations in the rehh packages in R [51]. For a better comparison of selection signals, |iHS| scores were transformed into −log10(2Φ(-|iHS|), in which Φ (x) is the cumulative Gaussian distribution function. Selection pressure on rice- and non-rice-infecting M. oryzae isolates at the individual population level was analyzed with the iHS test [52]. A resulting iHS greater than 0 with a p-value of ≤0.01 indicated positive selection. The XPEHH test was used to examine the comparative selection pressure between populations of rice- and non-rice-infecting isolates. The XPCLR [33] was used to detect and compare potential selective regions between rice- and non-rice-infecting populations. A resulting XPEHH value greater than 0 indicated rice-infecting M. oryzae population with selection pressure. On the contrary, values lower than 0 implied a selection in the non-rice infecting population. For the iHS and XPEHH tests, p-values were computed using the −log10 scale; thus, values greater than 2 were considered candidate selection sites. The top 1000 normalized XPCLR values for each window region in the XPCLR test represented a selection region.
2.4. Candidate Genes and Functional Annotation
Each SNP and sliding window region with genomic position and selective scores were calculated by the iHS, XPEHH, and XPCLR tests. According to the annotated reference genome file from the NCBI database, the potentially selective SNPs were remapped to the reference genomic genes. Amino acid sequences of candidate genes with significant selective signal SNPs were submitted to the Pathogen Host Interaction (PHI) database [53], the non-redundant (Nr) protein database (http://www.ncbi.nlm.nih.gov/ (accessed on 1 January 2021)), Gene Ontology (GO) (http://www.geneontology.org (accessed on 1 January 2021)), the Universal Protein Resourcce (UniProt) (https://www.uniprot.org (accessed on 1 January 2021)), Protein family (Pfam) database (http://pfam.xfam.org/ (accessed on 1 January 2021)), and eggnog database (http://eggnogdb.embl.de/ (accessed on 1 January 2021)) for functional prediction.
2.5. Effector Candidate Prediction
Gene amino acid sequences under selection were also submitted to SignalP-5.0 [54], TargetP 2.0 [55], and EffectorP [56] to predict potential effectors. SignalP-5.0 (http://www.cbs.dtu.dk/services/SignalP/ (accessed on 1 January 2021)) was adopted to predict the presence and location of signal peptide cleavage sites. The subcellular localizations of putative candidate effector proteins identified in this study were predicted with TargetP 2.0 (http://www.cbs.dtu.dk/services/TargetP/ (accessed on 1 January 2021)). EffectorP 2.0 was trained to detect the effector candidates in fungi. As the threshold to filter the obscure genes predicted, a 0.6 probability was used.
2.6. Host Directional Mutation
The amino acid sequence of selection associated gene MGG_13871 was extracted from each M. oryzae genome. Multiple sequence alignment was implemented in the ClusterW program in MEGA X [57]. Multiple sequence alignment of MGG_13871 amino acid sequences from rice- and non-rice-infecting populations was visualized by the ggmsa package in R (https://github.com/YuLab-SMU/ggmsa (accessed on 1 January 2021)).
3. Results
3.1. Population Genomic Divergence Driven by Host Adaptation in the M. oryzae Species Complex
To evaluate the dynamics in the genomic structures of the 197 M. oryzae isolates collected from 10 different hosts, we assessed the impacts of divergent genomic features on the adaptation of M. oryzae isolates to different hosts. We identified a total of 366,502 bi-allelic SNP loci in the exon regions within genomes. Phylogenetic analysis, principal component analysis (PCA), and population structure component analysis unraveled the relationship between different host-infecting M. oryzae populations. These analyses revealed significant differences between populations of M. oryzae isolates from different host plants. The evolutionary trend analyses revealed that the 197 M. oryzae isolates obtained from the 10 different hosts can be clustered in two major clades, with the first clade (clade-1) comprising rice-infecting isolates and the second clade (clade-2) containing non-rice-infecting isolates (Figure 1A). Moreover, higher bootstrap values were obtained for isolates from common host plants than isolates from distant host plants.
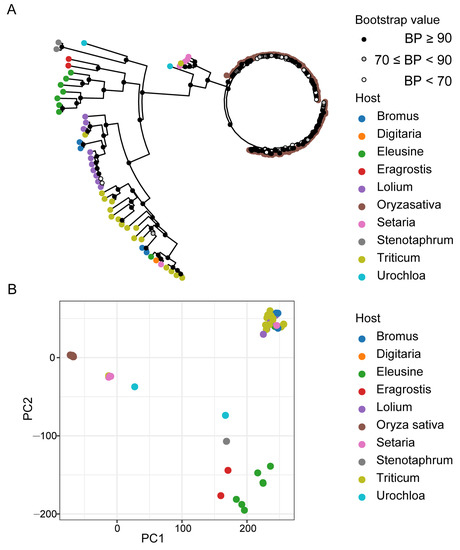
Figure 1.
Phylogenetic relationship of M. oryzae isolates from different host plants. (A) Neighbor-joining phylogenetic tree based on the whole-genome exon region single-nucleotide polymorphism (SNP) data from 197 isolates among 10 different hosts. The dot in white, grey, and black (nodes) indicates bootstrap support greater than 90%, between 70% and 90%, and lower than 70% after 1000 replications, respectively. (B) Two dimensions principal component analysis (PCA) showing the relationship of different hosts infesting M. oryzae populations. Colorful dots at the tips of branches mark the isolates from a variety of hosts.
We also observed a lesser genetic distance within clades consisting of rice-infecting isolate clades compared to clades containing isolates capable of infecting multiple hosts. The topological pattern obtained from the phylogenetic analysis further showed the aggregation of selected isolates from rice-infecting population with lesser evolutionary separation into distinct clusters, even under principal components 1 and 2 (PC1 and PC2) resolutions. The isolates adapted to non-rice hosts showed distant and dispersed topological distribution patterns (Figure 1B).
We examined the optimal number of clusters with the discriminant analysis of principal components (DAPC) based on the lowest associated Bayesian information criterion [58] and linear discriminants analysis. These analyses showed that the best number of clusters unambiguously correlated with the host preference of individual isolates within distinct populations (Figure 2A,B). However, cluster number adjustments resulted in the formation of marginal groups, with rice-infecting populations and non-rice-infecting populations having two groups each. The first group of the non-rice-infecting population predominantly consisted of isolates from Bromus spp., Digitaria spp., Lolium spp., and Triticum spp., while the other non-rice-infecting population comprised isolates that infect Eleusine spp., Eragrostis spp., and Stenotaphrum secundatum (Figure 2C). Regardless, an admixture of genetic signatures was present among the distinct M. oryzae populations.
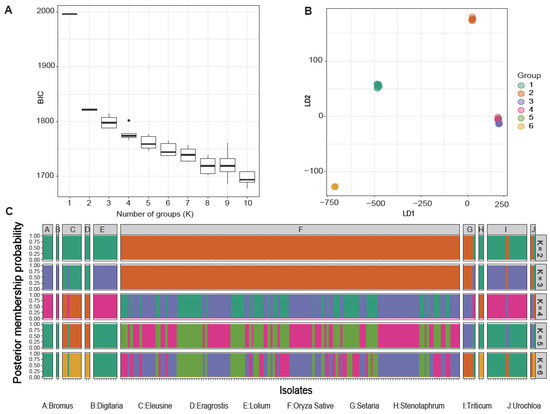
Figure 2.
Analysis of the population genetics of Magnaporthe oryzae according to the discriminant analysis of principal components (DAPC). (A) The Bayesian information criterion (BIC) distribution. (B) Scatter plot of the first (LD1) and second (LD2) linear discriminant functions with group numbers from 2 to 6. Dots with different colors represent each sample that was assigned to a designated group. (C) Bar plots of the posterior membership probabilities acquired from the DAPC analysis with K from 2 to 6. Each isolate is represented by one bar.
3.2. Selective Sweep Signatures in the Genomic Sequences of M. oryzae Populations
To exhaustively mine the selective region of beneficial genes, we performed iHS, XPEHH, and XPCLR analyses to identify the candidate region by scanning the whole genome within and between populations. In terms of population structure, we assumed that natural selection acted differently on the rice- and non-rice-infecting M. oryzae populations in accordance with their host differences. Further examination of individual SNPs or window regions with selection signals from three independent tests revealed a nearly uniform distribution of the selective regions across each chromosome (Figure 3). Although most of the SNPs and window regions identified within the respective populations’ genomes were under neutral selection, outliers in each test were detected that were considered the selective sweep region. A single population-based iHS analysis revealed 263 and 412 genes with −log10 p-values of >2 in rice- and non-rice-infecting isolate populations, respectively, along with 1277 SNP sites that mapped to the exon regions of 97 genes (Supplementary Tables S2 and S3). Similarly, XPEHH analyses between rice- and non-rice-infecting populations identified 6632 SNPs located in the exon regions of 102 genes with −log10 p-values >2 (Supplementary Table S4). Furthermore, whole-genome sliding window analysis records obtained from XPCLR tests identified a total of 321 genes as the top 1000 normalized cross-population composite likelihood ratios between the two populations (Supplementary Table S5).
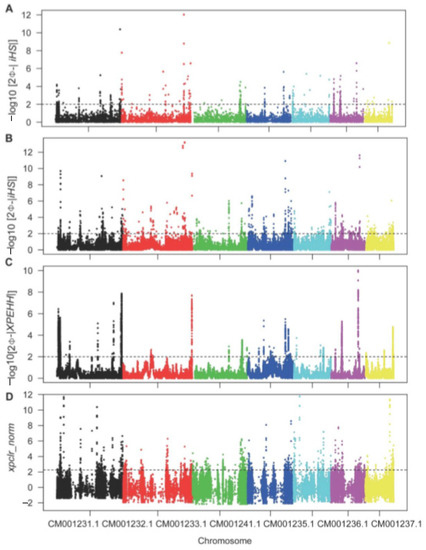
Figure 3.
Scatter plot of genome-wide selective signals in the integrated haplotype score (iHS) and the cross-population extended haplotype homozygosity (XPEHH) and cross-population composite likelihood ratio (XPCLR) tests. (A) Genome-wide distribution of -log10 p of standardized iHS in rice isolate population. (B) Genome-wide distribution of −log10 p of standardized iHS in non-rice isolate population. (C) Genome-wide distribution of −log10 p of standardized XPEHH scores between rice and non-rice isolate populations. (D) Genome-wide distribution of normalized XPCLR scores with 500 bp sliding windows between rice and non-rice isolate populations.
3.3. Selective Genes Associated with Host Speciation—Functional Annotation
We performed comparative functional annotation analyses of genes located within selective sweep regions on Nr, Pfam, Uniprot, eggnog, and PHI platforms to retrieve selection-associated genes. Although most of the genes recovered from selection prone regions of the genome have not been functionally characterized in rice blast fungus, our investigations revealed that 14 pathogenicity or virulence-associated genes, including the effector protein (Avr-Pita1) coding gene (MGG_15370) [59], had undergone positive selection in the non-rice-infecting population (XPEHH = −2.77) (Table 1). Positive selection of genes (MGG_07528 and MGG_07312) is likely an essential genetic phenomenon that drives host-shift among non-rice-infecting populations (iHS = 2.83 and iHS = 3.19, respectively).

Table 1.
Significantly selected pathogenicity-related genes identified in M.oryzae through iHS, XPEHH, and XPCLR analyses. PHI, Pathogen–Host Interaction database.
Furthermore, we observed that in the rice-infecting population, positive selection acted upon MGG_04842 along with genes for pathogenicity and survival of the rice blast fungus (iHS = 2.92). Additionally, functional analyses showed that pathogenicity-related genes, including MGG_14061, MGG_05464, MGG_10730, MGG_14767, MGG_02916, and MGG_17278, were positively selected in both rice- and non-rice-infecting populations (Table 1). These investigations also showed that seven members of glycosyl hydrolase family encoded by MGG_09738, MGG_09608, MGG_02911, MGG_00050, MGG_05489, MGG_16377, and MGG_07101, along with mixed-linked glucanase (MGG_07306) [60,61], cell wall glycosyl hydrolase (MGG_04305) [62], and cutinase (MGG_11091) [63], are positively selected exclusively in non-rice-infecting population.
3.4. Candidate Effectors Experiencing Positive Selection
Effectors play a key role in host identification for M. oryzae, so it is essential to discover potential effectors harbored in the selective sweep region. Effector candidates were predicted within genes selected with SignalP 5.0, TargetP 2.0, and EffectorP2.0 based on amino acid sequences. Integrated prediction analysis identified 20 candidate effector proteins with probability scores above 0.6 (Table 2). Remarkably, only candidates MGG_17666, MGG_15458, MGG_05538, MGG_00230, MGG_07352, and MGG_15370 showed positive selection status in non-rice-populations with iHS values of 2.63, 3.10, 3.91, 3.19, and 3.13 and an XPEHH score of −2.77, respectively. For rice-infecting population, candidates MGG_08214, MGG_07993, MGG_06231, MGG_16925, MGG_15106, MGG_07246, MGG_16953, and MGG_14374 were under positive selection with an iHS of 5.15; XPCLR values of 2.26, 2.71, 2.70, 3.45, 2.73, and 2.31; and an XPEHH score of 3.08, respectively (Table 2). Meanwhile, MGG_06234, MGG_16938, and MGG_07311 were selected for the rice- and non-rice-infecting populations. Additionally, diversified selection acted on the genes shared between rice- and non-rice-infecting populations. We also found that a hypothetical protein lacking the secretion signal motif selective gene (MGG_13871) experienced differential selection pressure in rice- and non-rice-infecting isolates. For instance, it was positively selected in the non-rice-infecting population (XPEHH = −3.14) but negatively selected in rice-infecting M. oryzae population (iHS = −2.97).
Table 2.
Dynamic characteristics of putative effector proteins identified in selective regions of the genomes.
3.5. MGG_13871 Experienced Host Directional Mutation
Furthermore, to uncover the fixed mutations in genes associated with specific phenotypes that contributed to population differentiation under natural selection, we further extracted the amino acid sequence of MGG_13871 from each genome of M. oryzae isolates in this study, because MGG_13871 underwent divergent selection in the rice- and non-rice-infecting M. oryzae populations. Multiple alignments of MGG_13871 amino acid sequences displayed host-directional mutations in two residues (Figure 4). At the 44th amino acid residue position, the amino acid was asparagine (N) in the rice-infecting population, but aspartic acid (D) in the non-rice-infecting population. Likewise, the lysine (K) in the rice-infecting population was substituted with lysine (K) in the non-rice-infecting population at the 102nd position. Taken together, we concluded that MGG_13871 experienced host-directed mutation in rice- and non-rice-infecting populations.
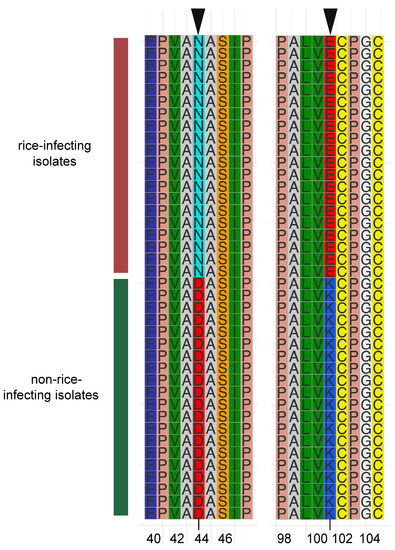
Figure 4.
Multiple amino acid sequence alignment of the selective motif of a major positively selected gene (MGG_13871) in non-rice-infecting isolates. Motif sequences of the selectable regions of 20 isolates each from rice- and non-rice-infecting M. oryzae were extracted for comparative motif sequence analysis. The red and green vertical bars represent the rice and non-rice host isolates, respectively. The arrows on the top indicate the directed mutation residues. The number at the bottom indicates the mutation’s position in the gene.
4. Discussion
In this study, whole-genome SNPs were used to resolve the population structure and composition in M. oryzae populations derived from 10 various host plants, including rice, wheat, and grass. This study further confirmed previous research, in that reshuffling pathotypes from multiple lineages favored the formation of identical isolates with no common cryptic lineages [8]. Host specificity is a noticeable genetic characteristic that has been the focus of whole-genome sequencing and multi-locus analysis in previous studies to decipher the genomic parameters of the host diversity of isolates within the M. oryzae species complex [17,21].
This study demonstrated that rice-infecting M. oryzae isolates constitute a monophyletic clade with closer evolutionary distance than non-rice hosts. These results partly showed that rice-infecting isolates are under an almost uniform selection pressure from rice hosts due to limited genetic diversity or genomic reshuffling among rice cultivars. Furthermore, we asserted that rice-infecting isolates originated from a recent common ancestor through asexual reproduction. However, a small number of isolates within the non-rice-infecting population with an identical host form unique discrete clusters with a relatively wider phylogenetic distance. We deduced that more genetic diversity in the non-rice hosts exerts tremendous selection pressure or induces higher incidences of gene flow among non-rice-infecting isolates, especially in clusters containing mixtures of isolates from Bromus spp. and Lolium spp. These observations support previous research suggesting that genetic exchange is a crucial contributor to the minimization of divergence between lineage-specific Magnaporthe isolates from non-rice hosts [8].
Additionally, the population structure analysis conducted in this study showed that wheat-infecting isolates belong to the M. oryzae species complex. These findings further confirm previous positions that the divergence of wheat blast isolates does not qualify them as new species. Although rice-infecting isolates are phylogenetically distant from Magnaporthe isolates from other hosts, they share relatively closer evolutionary ties with isolates from Setaria [17]. Herein, we demonstrated the grouping of Magnaporthe isolates into two distinct groups: rice-infecting populations and non-rice-infecting populations [64].
Furthermore, the evaluation of gene flow patterns in rice-infecting M. oryzae populations within Asia identified mainland China as the primary source of gene flow. We also observed that genetic material of M. oryzae populations from mainland China and Japan possibly first converged in Taiwan, China, and later served as the gene pool for Korean isolates (Supplementary Figure S1). This may be attributed to the fact that mainland China is the origin of wild and domesticated rice [65]. We posited that rice-infecting Magnaporthe isolates possibly evolved along with their rice host in mainland China and were later introduced to subsequent regions, including Taiwan, China, and Korea. Although the gene flow was from mainland China to Taiwan, isolates from Taiwan display relatively greater evolutionary distance from isolates sampled from the neighboring Fujian province in China [64]. For the rice-infecting isolates, rice varieties also have a significant impact on M. oryzae population structure. Most of the isolates are from Japonica growing areas such as Taiwan, the north of China, and Japan, which are close to one another. In contrast, isolates sampled from indica growing areas, including the Fujian and Yunan provinces in China and India, frequently assembled into the same cluster (Supplementary Figure S2).
Host shift, host jump, and host expansion are adaptation characteristics that allow M. oryzae to successfully co-evolve to match agronomic changes (blast resistance) associated with the domestication of rice [4]. Previous studies have speculatively proposed that the blast fungus possibly undergoes accelerated genome reshuffling to support adaptive evolution, which enables it to experience host shift, host jump, or host expansion [22,24]. We identified, for the first time, credible selection-prone genomic regions containing protein-coding genes in rice- and non-rice-infecting populations through the integrated application of the iHS, XPEHH, and XPCLR tests. To eliminate bias resulting from high-frequency transposons in the genome of Magnaporthe spp., we only considered and compared mutations occurring in the exonic regions of individual isolates within the respective populations. Generally, pathogens retain survival or beneficial mutations during host adaptation induced speciation. Therefore, positively selected genes likely include genes that play crucial roles in the progression of host–pathogen interactions [66]. Effector proteins are small secreted proteins from fungal pathogens that have been shown to play dynamic roles in the progression of host–pathogen interactions by either facilitating host identification or acting as pathogenicity and/or virulence factors [67]. Additional studies have shown that candidate effector proteins in blast fungus undergo rapid evolution to yield new pathotypes of M. oryzae with broader host ranges or adaptations for specific hosts [68].
We showed that 17 candidate effector proteins undergo differential selection sweep in rice- and non-rice-infecting M. oryzae populations and speculatively concluded that the differential selection sweep of pathogenesis-related candidate effector proteins within and between rice- and non-rice-infecting M. oryzae populations likely accounts for host divergence and host specificity among Magnaporthe spp. We also showed that selection-prone genes, including MGG_13871 and MGG_07815, experience differential host-specific mutations between rice- and non-rice-infecting populations. These two candidate selectable genes experienced uniform and synonymous amino acid substitution in rice- and non-rice-infecting populations. The site, amino acid, and type of selection pressure experienced by MGG_13871 and MGG_07815 varied between the two populations. For instance, MGG_13871 experienced positive selection in non-rice-infecting populations and negative selection in rice-infecting populations. We accordingly concluded that MGG_13871 likely constitutes a host specificity determiner in Magnaporthe spp. [69].
Meanwhile, specific selection prone candidate effectors in rice- and non-rice-infecting populations, including MGG_17666, MGG_15458, and MGG_08435, could also play additional roles in modulating population differentiation and host specificity between the distinct Magnaporthe populations reported in this study. Moreover, we also identified a cutinase gene, MGG_11091, in the selective sweep region, an essential enzyme required to degrade the host cuticle to support the successful invasion of blast fungus into host tissues. MGG_05489 and MGG_16377 experienced positive selection in rice-infecting populations. These two genes are members of the glycosyl hydrolase family that were previously implicated in the pathogenesis of Magnaporthe spp. However, when compared to rice-infecting isolates, wheat-infecting M. oryzae isolates seemingly favor the seed; therefore, positive selection of mixed-linked glucanase MGG_07306 is possibly a host adaptation mechanism acquired by M. oryzae.
In summary, our results provide the crucial insight that selective sweeping plays a role in driving M. oryzae population differentiation associated with host adaptation, and we revealed potential genomic regions of interest to understand the genetic mechanisms of host specialization in M. oryzae.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-2607/9/3/562/s1, Figure S1: Gene flow evaluation in Asia; Figure S2: Phylogenetic tree of M. oryzae; Table S1: M.oryzae isolates’ host information; Table S2: Integrated haplotype score (iHS) test in rice host M. oryzae isolates; Table S3: iHS test in non-rice host M. oryzae isolates; Table S4: Cross-population extended haplotype homozygosity (XPEHH) test between rice and non-rice host M. oryzae isolates; Table S5: Cross-population composite likelihood ratio (XPCLR) test between rice and non-rice host M.oryzae isolates.
Author Contributions
Z.W., W.T., and G.D. conceived and designed the research; G.D., J.B., and H.C. collected the data; W.T., G.D., and J.B. performed the bioinformatic and statistical analyses; X.C., J.X., and Y.L. helped with validation analyses; Z.W., W.T., G.D., and H.Z. wrote the manuscript; Z.W. and W.T. were primarily responsible for the final content and for editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. U1805232).
Acknowledgments
The authors are sincerely grateful to Justice Norvienyeku of the Fujian Agriculture and Forestry University (FAFU) for assisting with the revision of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic Structure and Diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.J. On the Trail of a Cereal Killer: Exploring the Biology of Magnaporthe grisea. Annu. Rev. Microbiol. 2003, 57, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.A.; Webster, R.K. Occurrence, Distribution, Epidemiology, Cultivar Reaction, and Management of Rice Blast Disease in California. Plant. Dis. 2001, 85, 1096–1102. [Google Scholar] [CrossRef]
- Kato, H.; Yamamoto, M.; Yamaguchi-Ozaki, T.; Kadouchi, H.; Iwamoto, Y.; Nakayashiki, H.; Tosa, Y.; Mayama, S.; Mori, N. Pathogenicity, Mating Ability and DNA Restriction Fragment Length Polymorphisms of Pyricularia Populations Isolated from Gramineae, Bambusideae and Zingiberaceae Plants. J. Gen. Plant. Pathol. 2000, 66, 30–47. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Attack of the Clones. Science 2012, 337, 636. [Google Scholar] [CrossRef]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.-R.; Pan, H.; et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [Google Scholar] [CrossRef]
- Gladieux, P.; Condon, B.; Ravel, S.; Soanes, D.; Maciel, J.L.N.; Nhani, A.; Chen, L.; Terauchi, R.; Lebrun, M.-H.; Tharreau, D.; et al. Gene Flow between Divergent Cereal- and Grass-Specific Lineages of the Rice Blast Fungus Magnaporthe oryzae. Mbio 2018, 9, e01219-17. [Google Scholar] [CrossRef]
- Fraser, H.B.; Levy, S.; Chavan, A.; Shah, H.B.; Perez, J.C.; Zhou, Y.; Siegal, M.L.; Sinha, H. Polygenic cis-regulatory adaptation in the evolution of yeast pathogenicity. Genome Res. 2012, 22, 1930–1939. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saleh, D.; Xu, P.; Shen, Y.; Li, C.; Adreit, H.; Milazzo, J.; RavignÉ, V.; Bazin, E.; NottÉGhem, J.-L.; Fournier, E.; et al. Sex at the origin: An Asian population of the rice blast fungus Magnaporthe oryzae reproduces sexually. Mol. Ecol. 2012, 21, 1330–1344. [Google Scholar] [CrossRef]
- Saleh, D.; Milazzo, J.; Adreit, H.; Fournier, E.; Tharreau, D. South-East Asia is the center of origin, diversity and dispersion of the rice blast fungus, Magnaporthe oryzae. New Phytol. 2014, 201, 1440–1456. [Google Scholar] [CrossRef]
- Igarashi, S. Update on wheat blast (Pyricularia oryzae) in Brazil. In Proceedings of the International Conference-Wheat for the Nontaditional Warm Areas, Foz Do Iguaçu, Brazil, 29 July–3 August 1990; pp. 480–483. [Google Scholar]
- Zaffarano, P.L.; McDonald, B.A.; Linde, C.C. Rapid speciation following recent host shifts in the plant pathogenic fungus Rhynchosporium. Evolution 2008, 62, 1418–1436. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.N.; Talhinhas, P.; Cai, L.E.I.; Manuel, L.; Gichuru, E.K.; Loureiro, A.; VÁRzea, V.; Paulo, O.S.; Batista, D. Host-jump drives rapid and recent ecological speciation of the emergent fungal pathogen Colletotrichum kahawae. Mol. Ecol. 2012, 21, 2655–2670. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, O.; Peever, T.L.; Chilvers, M.I.; Özkilinc, H.; Can, C.; Abbo, S.; Shtienberg, D.; Sherman, A. Ecological Genetic Divergence of the Fungal Pathogen Didymella rabiei on Sympatric Wild and Domesticated Cicer spp. (Chickpea). Appl. Environ. Microbiol. 2010, 76, 30. [Google Scholar] [CrossRef]
- Stukenbrock, E.H.; Banke, S.; Javan-Nikkhah, M.; McDonald, B.A. Origin and Domestication of the Fungal Wheat Pathogen Mycosphaerella graminicola via Sympatric Speciation. Mol. Biol. Evol. 2007, 24, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Couch, B.C.; Fudal, I.; Lebrun, M.H.; Tharreau, D.; Valent, B.; van Kim, P.; Notteghem, J.L.; Kohn, L.M. Origins of host-specific populations of the blast pathogen Magnaporthe oryzae in crop domestication with subsequent expansion of pandemic clones on rice and weeds of rice. Genetics 2005, 170, 613–630. [Google Scholar] [CrossRef]
- Hirata, K.; Kusaba, M.; Chuma, I.; Osue, J.; Nakayashiki, H.; Mayama, S.; Tosa, Y. Speciation in Pyricularia inferred from multilocus phylogenetic analysis. Mycol. Res. 2007, 111, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Yamagashira, A.; Iwai, C.; Misaka, M.; Hirata, K.; Fujita, Y.; Tosa, Y.; Kusaba, M. Taxonomic characterization of Pyricularia isolates from green foxtail and giant foxtail, wild foxtails in Japan. J. Gen. Plant. Pathol. 2008, 74, 230–241. [Google Scholar] [CrossRef]
- Yoshida, K.; Saunders, D.G.O.; Mitsuoka, C.; Natsume, S.; Kosugi, S.; Saitoh, H.; Inoue, Y.; Chuma, I.; Tosa, Y.; Cano, L.M.; et al. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genom. 2016, 17, 370. [Google Scholar] [CrossRef]
- Chiapello, H.; Mallet, L.; Guérin, C.; Aguileta, G.; Amselem, J.; Kroj, T.; Ortega-Abboud, E.; Lebrun, M.-H.; Henrissat, B.; Gendrault, A.; et al. Deciphering Genome Content and Evolutionary Relationships of Isolates from the Fungus Magnaporthe oryzae Attacking Different Host Plants. Genome Biol. Evol. 2015, 7, 2896–2912. [Google Scholar] [CrossRef]
- Wicker, T.; Oberhaensli, S.; Parlange, F.; Buchmann, J.P.; Shatalina, M.; Roffler, S.; Ben-David, R.; Doležel, J.; Šimková, H.; Schulze-Lefert, P.; et al. The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat. Genet. 2013, 45, 1092–1096. [Google Scholar] [CrossRef]
- Latorre, S.M.; Reyes-Avila, C.S.; Malmgren, A.; Win, J.; Kamoun, S.; Burbano, H.A. Differential loss of effector genes in three recently expanded pandemic clonal lineages of the rice blast fungus. BMC Biol. 2020, 18, 88. [Google Scholar] [CrossRef]
- Hyon, G.-S.; Nga, N.T.T.; Chuma, I.; Inoue, Y.; Asano, H.; Murata, N.; Kusaba, M.; Tosa, Y. Characterization of interactions between barley and various host-specific subgroups of Magnaporthe oryzae and M. grisea. J. Gen. Plant. Pathol. 2012, 78, 237–246. [Google Scholar] [CrossRef]
- Kohli, M.M.; Mehta, Y.R.; Guzman, E.; De Viedma, L.; Cubilla, L.E. Pyricularia Blast—A Threat to Wheat Cultivation. Czech. J. Genet. Plant. 2011, 47, S130–S134. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, W.; Lin, F.; Zhang, Y.; Yi, Y.; Wang, B.; Lu, G.; Wang, Z.; Wu, W. AVR1-CO39 Is a Predominant Locus Governing the Broad Avirulence of Magnaporthe oryzae 2539 on Cultivated Rice (Oryza sativa L.). Mol. Plant.-Microbe Interact. 2010, 24, 13–17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leong, S.A. The Ins and Outs of Host Recognition of Magnaporthe oryzae. In Genomics of Disease; Gustafson, J.P., Taylor, J., Stacey, G., Eds.; Springer: New York, NY, USA, 2008; pp. 199–216. [Google Scholar] [CrossRef]
- Tosa, Y.; Osue, J.; Eto, Y.; Oh, H.-S.; Nakayashiki, H.; Mayama, S.; Leong, S.A. Evolution of an Avirulence Gene, AVR1-CO39, Concomitant with the Evolution and Differentiation of Magnaporthe oryzae. Mol. Plant.Microbe Interact. 2005, 18, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Norvienyeku, J.; Chen, M.; Bao, J.; Lin, L.; Chen, L.; Lin, Y.; Wu, X.; Cai, Z.; Zhang, Q.; et al. Directional Selection from Host Plants Is a Major Force Driving Host Specificity in Magnaporthe Species. Sci. Rep. 2016, 6, 25591. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhong, Z.; Shi, M.; Zhang, L.; Lin, L.; Hong, Y.; Fang, T.; Zhu, Y.; Guo, J.; Zhang, L.; et al. Comparative genomic analysis revealed rapid differentiation in the pathogenicity-related gene repertoires between Pyricularia oryzae and Pyricularia penniseti isolated from a Pennisetum grass. BMC Genom. 2018, 19, 927. [Google Scholar] [CrossRef]
- Wright, S. The genetical structure of populations. Ann. Eugen 2012, 15, 323–354. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Gross, L. Clues to Our Past: Mining the Human Genome for Signs of Recent Selection. PLoS Biol. 2006, 4, e94. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.Q.; Pritchard, J.K. Correction: A map of recent positive selection in the human genome (vol 4, pg 446, 2006). PLoS Biol. 2007, 5, 1382. [Google Scholar] [CrossRef]
- Hartmann, F.E.; McDonald, B.A.; Croll, D. Genome-wide evidence for divergent selection between populations of a major agricultural pathogen. Mol. Ecol. 2018, 27, 2725–2741. [Google Scholar] [CrossRef]
- Mohd-Assaad, N.; McDonald, B.A.; Croll, D. Genome-Wide Detection of Genes Under Positive Selection in Worldwide Populations of the Barley Scald Pathogen. Genome Biol. Evol. 2018, 10, 1315–1332. [Google Scholar] [CrossRef]
- Derbyshire, M.C.; Denton-Giles, M.; Hane, J.K.; Chang, S.; Mousavi-Derazmahalleh, M.; Raffaele, S.; Buchwaldt, L.; Kamphuis, L.G. Selective sweeps in populations of the broad host range plant pathogenic fungus Sclerotinia sclerotiorum. bioRxiv 2018, 352930. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, H.; Zhang, Q.; Ding, X. Identification of Selection Footprints on the X Chromosome in Pig. PLoS ONE 2014, 9, e94911. [Google Scholar] [CrossRef]
- Taye, M.; Lee, W.; Caetano-Anolles, K.; Dessie, T.; Hanotte, O.; Mwai, O.A.; Kemp, S.; Cho, S.; Oh, S.J.; Lee, H.-K.; et al. Whole genome detection of signature of positive selection in African cattle reveals selection for thermotolerance. Anim. Sci. J. 2017, 88, 1889–1901. [Google Scholar] [CrossRef]
- He, Q.; Kim, K.-W.; Park, Y.-J. Population genomics identifies the origin and signatures of selection of Korean weedy rice. Plant. Biotechnol. J. 2017, 15, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Prevosti, A.; Ocaña, J.; Alonso, G. Distances between populations ofDrosophila subobscura, based on chromosome arrangement frequencies. Theor. Appl. Genet. 1975, 45, 231–241. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.P.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef]
- Gautier, M.; Klassmann, A.; Vitalis, R. REHH 2.0: A reimplementation of the R package REHH to detect positive selection from haplotype structure. Mol. Ecol. Resour. 2016, 17, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Naves, M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 2011, 20, 3128–3143. [Google Scholar] [CrossRef]
- Winnenburg, R.; Urban, M.; Beacham, A.; Baldwin, T.K.; Holland, S.; Lindeberg, M.; Hansen, H.; Rawlings, C.; Hammond-Kosack, K.E.; Köhler, J. PHI-base update: Additions to the pathogen host interaction database. Nucleic Acids Res. 2008, 36, D572–D576. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. EffectorP: Predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2016, 210, 743–761. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Chuma, I.; Isobe, C.; Hotta, Y.; Ibaragi, K.; Futamata, N.; Kusaba, M.; Yoshida, K.; Terauchi, R.; Fujita, Y.; Nakayashiki, H.; et al. Multiple Translocation of the AVR-Pita Effector Gene among Chromosomes of the Rice Blast Fungus Magnaporthe oryzae and Related Species. PLoS Pathog. 2011, 7, e1002147. [Google Scholar] [CrossRef]
- Martin, K.; McDougall, B.M.; McIlroy, S.; Chen, J.; Seviour, R.J. Biochemistry and molecular biology of exocellular fungal β-(1,3)- and β-(1,6)-glucanases. Fems Microbiol. Rev. 2007, 31, 168–192. [Google Scholar] [CrossRef]
- Mouyna, I.; Hartl, L.; Latgé, J.-P. β-1,3-glucan modifying enzymes in Aspergillus fumigatus. Front. Microbiol. 2013, 4, 81. [Google Scholar] [CrossRef]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell Wall Hydrolases in Bacteria: Insight on the Diversity of Cell Wall Amidases, Glycosidases and Peptidases Toward Peptidoglycan. Front. Microbiol. 2019, 10, 331. [Google Scholar] [CrossRef]
- Sweigard, J.A.; Chumley, F.G.; Valent, B. Cloning and analysis of CUT1, a cutinase gene from Magnaporthe grisea. Mol. Gen. Genet. Mgg 1992, 232, 174–182. [Google Scholar] [CrossRef]
- Zhong, Z.; Chen, M.; Lin, L.; Han, Y.; Bao, J.; Tang, W.; Lin, L.; Lin, Y.; Somai, R.; Lu, L.; et al. Population genomic analysis of the rice blast fungus reveals specific events associated with expansion of three main clades. ISME J. 2018, 12, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Gutaker, R.M.; Groen, S.C.; Bellis, E.S.; Choi, J.Y.; Pires, I.S.; Bocinsky, R.K.; Slayton, E.R.; Wilkins, O.; Castillo, C.C.; Negrão, S.; et al. Genomic history and ecology of the geographic spread of rice. Nat. Plants 2020, 6, 492–502. [Google Scholar] [CrossRef]
- Tiffin, P.; Moeller, D.A. Molecular evolution of plant immune system genes. Trends Genet. Tig. 2006, 22, 662–670. [Google Scholar] [CrossRef]
- Sánchez-Vallet, A.; Fouché, S.; Fudal, I.; Hartmann, F.E.; Soyer, J.L.; Tellier, A.; Croll, D. The Genome Biology of Effector Gene Evolution in Filamentous Plant Pathogens. Annu. Rev. Phytopathol. 2018, 56, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-T.; Ko, J.; Song, H.; Choi, G.; Kim, H.; Jeon, J.; Cheong, K.; Kang, S.; Lee, Y.-H. Evolution of the Genes Encoding Effector Candidates Within Multiple Pathotypes of Magnaporthe oryzae. Front. Microbiol. 2019, 10, 2575. [Google Scholar] [CrossRef] [PubMed]
- Quibod, I.L.; Perez-Quintero, A.; Booher, N.J.; Dossa, G.S.; Grande, G.; Szurek, B.; Vera Cruz, C.; Bogdanove, A.J.; Oliva, R. Effector Diversification Contributes to Xanthomonas oryzae pv. oryzae Phenotypic Adaptation in a Semi-Isolated Environment. Sci. Rep. 2016, 6, 34137. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).