
Fecal Microbiome and Resistome Profiling of Healthy and Diseased Pakistani Individuals Using Next-Generation Sequencing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Sample Collection
2.3. Extraction, Quantification and Normalization of Genomic DNA
2.4. NGS Libraries Preparation
2.5. NGS Bioinformatics Analysis
3. Results
3.1. Selection of Samples for Shotgun Metagenome Analysis
3.2. Shotgun Metagenome Sequencing
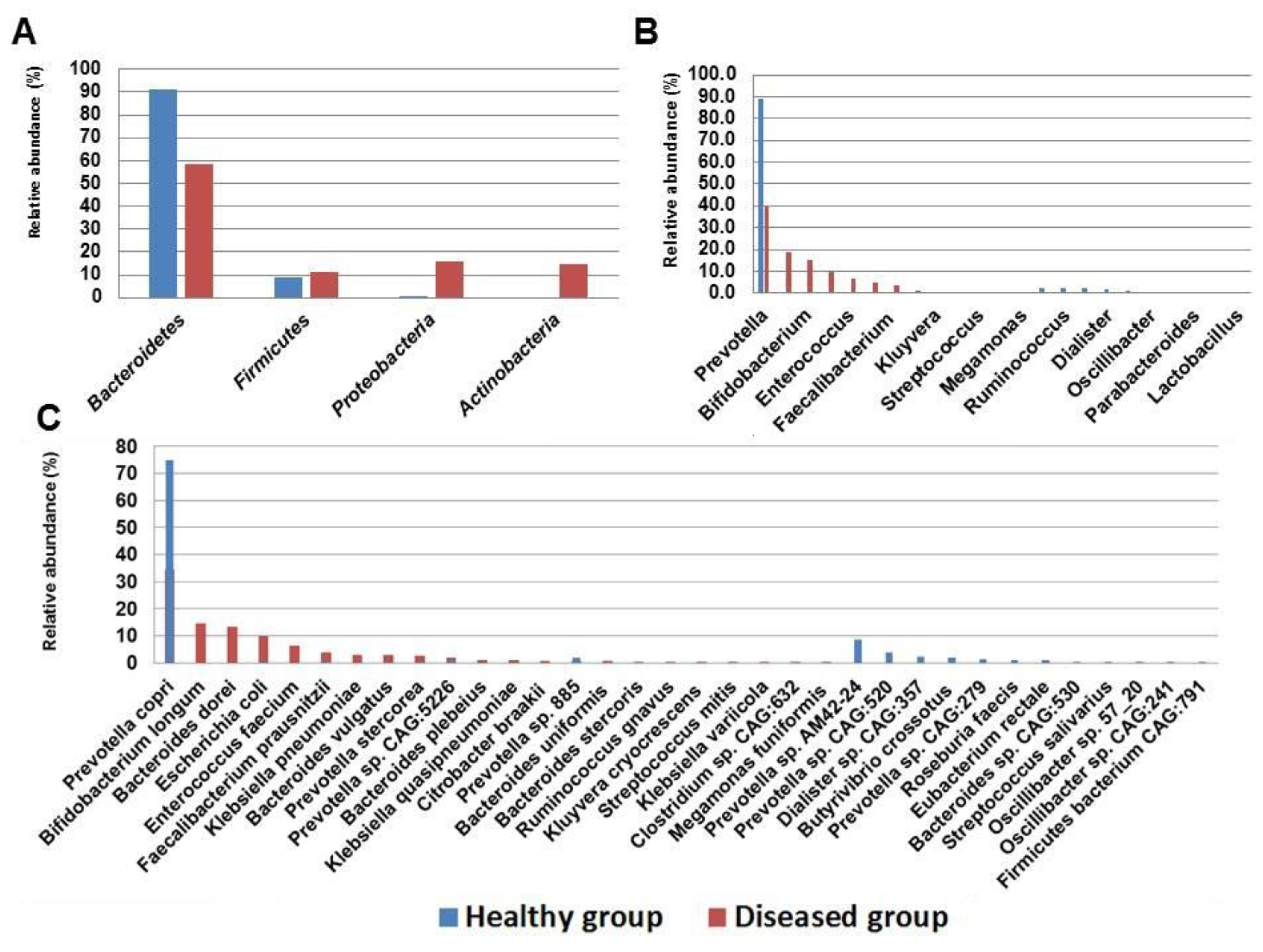
3.3. Bacterial Profiling of Fecal Microbiome of Healthy Subjects at Various Levels
3.4. Bacterial Profiling of Fecal Microbiome of Diseased Subjects at Various Levels
3.5. Comparative Bacterial Profiling of Healthy and Diseased Subjects
3.6. NGS-Based Resistome Analysis
3.7. Resistome of Healthy Subjects
3.8. Resistome of Diseased Subjects
3.9. Comparative Resistome Analysis
4. Discussion
5. Conclusions and Future Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Willmann, M.; Vehreschild, M.J.; Biehl, L.M.; Vogel, W.; Dörfel, D.; Hamprecht, A.; Seifert, H.; Autenrieth, I.B.; Peter, S. Distinct impact of antibiotics on the gut microbiome and resistome: A longitudinal multicenter cohort study. BMC Biol. 2019, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.O.; Dantas, G.; Church, G.M. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009, 325, 1128–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, D.; Langdon, A.; Dantas, G. Understanding the impact of antibiotic perturbation on the human microbiome. Genome Med. 2020, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Casals-Pascual, C.; Vergara, A.; Vila, J. Intestinal microbiota and antibiotic resistance: Perspectives and solutions. Hum. Microbiome J. 2018, 9, 11–15. [Google Scholar] [CrossRef]
- Duan, H.; Yu, L.; Tian, F.; Zhai, Q.; Fan, L.; Chen, W. Antibiotic-induced gut dysbiosis and barrier disruption and the potential protective strategies. Crit. Rev. Food Sci. Nutr. 2020, 1–26. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [Green Version]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.J.; Hasan, M.N.; Azhar, E.I.; Yasir, M. Association of gut dysbiosis with intestinal metabolites in response to antibiotic treatment. Hum. Microbiome J. 2019, 11, 100054. [Google Scholar] [CrossRef]
- Kho, Z.Y.; Lal, S.K. The human gut microbiome–a potential controller of wellness and disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef] [Green Version]
- Francino, M. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, F.; Moitinho-Silva, L.; Blasche, S.; Jahn, M.T.; Gossmann, T.I.; Huerta-Cepas, J.; Hercog, R.; Luetge, M.; Bahram, M.; Pryszlak, A. Antibiotics-induced monodominance of a novel gut bacterial order. Gut 2019, 68, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.; Bayer, F.; Pfrunder-Cardozo, K.R.; Buckling, A.; Hall, A.R. Resident microbial communities inhibit growth and antibiotic-resistance evolution of Escherichia coli in human gut microbiome samples. PLoS Biol. 2020, 18, e3000465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekmekciu, I.; Von Klitzing, E.; Fiebiger, U.; Escher, U.; Neumann, C.; Bacher, P.; Scheffold, A.; Kühl, A.A.; Bereswill, S.; Heimesaat, M.M. Immune responses to broad-spectrum antibiotic treatment and fecal microbiota transplantation in mice. Front. Immunol. 2017, 8, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, S.; Mian, F.M.; Stanisz, A.M.; Bindels, L.B.; Cambier, E.; Ben-Amram, H.; Koren, O.; Forsythe, P.; Bienenstock, J. Low-dose penicillin in early life induces long-term changes in murine gut microbiota, brain cytokines and behavior. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Forslund, K.; Sunagawa, S.; Kultima, J.R.; Mende, D.R.; Arumugam, M.; Typas, A.; Bork, P. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013, 23, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yang, X.; Qin, J.; Lu, N.; Cheng, G.; Wu, N.; Pan, Y.; Li, J.; Zhu, L.; Wang, X. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, W.; Liang, S.; Yamasaki, S.; Chen, X.; Shi, L.; Ye, L. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.; Vaz-Moreira, I.; Tamames, J.; Manaia, C.M.; Martínez, J.L. Metagenomic analysis of an urban resistome before and after wastewater treatment. Sci. Rep. 2020, 10, 1–9. [Google Scholar]
- Waseem, H.; Ali, J.; Sarwar, F.; Khan, A.; Rehman, H.S.U.; Choudri, M.; Arif, N.; Subhan, M.; Saleem, A.R.; Jamal, A. Assessment of knowledge and attitude trends towards antimicrobial resistance (AMR) among the community members, pharmacists/pharmacy owners and physicians in district Sialkot, Pakistan. Antimicrob. Resist. Infect. Control 2019, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Afridi, O.K.; Ali, J.; Chang, J.H. Next-Generation Sequencing Based Gut Resistome Profiling of Broiler Chickens Infected with Multidrug-Resistant Escherichia coli. Animals 2020, 10, 2350. [Google Scholar] [CrossRef]
- Hayat, K.; Rosenthal, M.; Gillani, A.H.; Zhai, P.; Aziz, M.M.; Ji, W.; Chang, J.; Hu, H.; Fang, Y. Perspective of Pakistani physicians towards hospital antimicrobial stewardship programs: A multisite exploratory qualitative study. Int. J. Environ. Res. Public Health 2019, 16, 1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsin, M.; Van Boeckel, T.P.; Saleemi, M.K.; Umair, M.; Naseem, M.N.; He, C.; Khan, A.; Laxminarayan, R. Excessive use of medically important antimicrobials in food animals in Pakistan: A five-year surveillance survey. Glob. Health Action 2019, 12, 1697541. [Google Scholar] [CrossRef] [Green Version]
- MacFaddin, J. Biochemical Tests for Identification of Medical Bacteria; Williams and Wilkins: Philadelphia, PA, USA, 2000; p. 113. [Google Scholar]
- Weinstein, M.P. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Annapolis Junction, MD, USA, 2019. [Google Scholar]
- Mirsepasi, H.; Persson, S.; Struve, C.; Andersen, L.O.; Petersen, A.M.; Krogfelt, K.A. Microbial diversity in fecal samples depends on DNA extraction method: easyMag DNA extraction compared to QIAamp DNA stool mini kit extraction. BMC Res. Notes 2014, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Hunt, M.; Mather, A.E.; Sánchez-Busó, L.; Page, A.J.; Parkhill, J.; Keane, J.A.; Harris, S.R. ARIBA: Rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genom. 2017, 3, e000131. [Google Scholar] [CrossRef]
- De, R. Metagenomics: Aid to combat antimicrobial resistance in diarrhea. Gut Pathog. 2019, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.S.R.; Stärk, K.D.; Munk, P.; Leekitcharoenphon, P.; Bossers, A.; Luiken, R.; Sarrazin, S.; Lukjancenko, O.; Pamp, S.J.; Bortolaia, V. Addressing learning needs on the use of metagenomics in antimicrobial resistance surveillance. Front. Public Health 2020, 8, 38. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, Z.; Tan, L.; Wang, X.; Xue, Y.; Wang, S.; Wang, Q.; Das, R.; Lin, H.; Hou, J. Gut resistomes, microbiota and antibiotic residues in Chinese patients undergoing antibiotic administration and healthy individuals. Sci. Total Environ. 2020, 705, 135674. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [Green Version]
- Rampelli, S.; Soverini, M.; D’Amico, F.; Barone, M.; Tavella, T.; Monti, D.; Capri, M.; Astolfi, A.; Brigidi, P.; Biagi, E. Shotgun metagenomics of gut microbiota in humans with up to extreme longevity and the increasing role of xenobiotic degradation. Msystems 2020, 5, e00124-20. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, E.D.; Sonnenburg, J.L. Starving our microbial self: The deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014, 20, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, C.F.; Haiser, H.J.; Turnbaugh, P.J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 2013, 152, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.U.; Khan, F.U.; Hayat, K.; Chang, J.; Saeed, A.; Khan, Z.; Ashraf, M.; Rasheed, U.M.; Atif, N.; Ji, W. Knowledge, attitude and practices among consumers toward antibiotics use and antibiotic resistance in Swat, Khyber-Pakhtunkhwa, Pakistan. Expert Rev. Anti-Infect. Ther. 2020, 18, 937–946. [Google Scholar] [CrossRef]
- Saleem, Z.; Hassali, M.A.; Godman, B.; Fatima, M.; Ahmad, Z.; Sajid, A.; Rehman, I.U.; Nadeem, M.U.; Javaid, Z.; Malik, M. Sale of WHO AWaRe groups antibiotics without a prescription in Pakistan: A simulated client study. J. Pharm. Policy Pract. 2020, 13, 1–8. [Google Scholar] [CrossRef]
- Klein, E.Y.; Van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, E3463–E3470. [Google Scholar] [CrossRef] [Green Version]
- Hayat, K.; Rosenthal, M.; Gillani, A.H.; Chang, J.; Ji, W.; Yang, C.; Jiang, M.; Zhao, M.; Fang, Y. Perspective of key healthcare professionals on antimicrobial resistance and stewardship programs: A multicenter cross-sectional study from Pakistan. Front. Pharmacol. 2020, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Buelow, E.; Gonzalez, T.B.; Versluis, D.; Oostdijk, E.A.; Ogilvie, L.A.; van Mourik, M.S.; Oosterink, E.; van Passel, M.W.; Smidt, H.; D’Andrea, M.M. Effects of selective digestive decontamination (SDD) on the gut resistome. J. Antimicrob. Chemother. 2014, 69, 2215–2223. [Google Scholar] [CrossRef]
- Khan, M.; Siddiqui, S.; Haider, S.; Zafar, A.; Zafar, F.; Khan, R.; Afshan, K.; Jabeen, A.; Khan, M.; Hasan, R. Infection control education: Impact on ventilator-associated pneumonia rates in a public sector intensive care unit in Pakistan. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Nahid, F.; Khan, A.A.; Rehman, S.; Zahra, R. Prevalence of metallo-β-lactamase NDM-1-producing multi-drug resistant bacteria at two Pakistani hospitals and implications for public health. J. Infect. Public Health 2013, 6, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Rettedal, E.A.; Van Der Helm, E.; Ellabaan, M.; Panagiotou, G.; Sommer, M.O. Antibiotic treatment drives the diversification of the human gut resistome. Genom. Proteom. Bioinform. 2019, 17, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, B.; Jiang, X.; Yang, Y.; Wells, G.F.; Zhang, T.; Li, X. Antibiotic resistome in a large-scale healthy human gut microbiota deciphered by metagenomic and network analyses. Environ. Microbiol. 2018, 20, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.J. The structure and diversity of human, animal and environmental resistomes. Microbiome 2016, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tariq, A.; Haque, A.; Ali, A.; Bashir, S.; Habeeb, M.A.; Salman, M.; Sarwar, Y. Molecular profiling of antimicrobial resistance and integron association of multidrug-resistant clinical isolates of Shigella species from Faisalabad, Pakistan. Can. J. Microbiol. 2012, 58, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Faizullah, M.; Rahman, N.; Umar, M.; Anwar, M.; Sarfraz, M. A cross-sectional study on knowledge, attitude and practices of medical doctors towards antibiotic prescribing patterns and resistance in Khyber Pakhtun Khawah, Pakistan. J. Appl. Pharm. Sci. 2017, 7, 38–46. [Google Scholar]
- English, B.K.; Gaur, A.H. The use and abuse of antibiotics and the development of antibiotic resistance. Hot Top. Infect. Immun. Child. 2010, VI, 73–82. [Google Scholar]
- Radhakrishnan, R.; Rajesh, J.; Dinesh, N.; Thangavelu, C.; Sankaran, K. High-throughput method for Antibiotic Susceptibility testing based on fluorescein Quenching by Bacteria: Application to Urinary tract infection. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, G.A.; Berglund, B.; Khan, K.M.; Lindgren, P.-E.; Fick, J. Occurrence and abundance of antibiotics and resistance genes in rivers, canal and near drug formulation facilities–a study in Pakistan. PLoS ONE 2013, 8, e62712. [Google Scholar] [CrossRef]
- Tsigalou, C.; Konstantinidis, T.; Stavropoulou, E.; Bezirtzoglou, E.E.; Tsakris, A. Potential Elimination of Human Gut Resistome by Exploiting the Benefits of Functional Foods. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vester-Andersen, M.; Mirsepasi-Lauridsen, H.; Prosberg, M.; Mortensen, C.; Träger, C.; Skovsen, K.; Thorkilgaard, T.; Nøjgaard, C.; Vind, I.; Krogfelt, K.A. Increased abundance of proteobacteria in aggressive Crohn’s disease seven years after diagnosis. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.T.; Amos, G.C.; Murphy, A.R.; Murch, S.; Wellington, E.M.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1–8. [Google Scholar] [CrossRef]
- Pilmis, B.; Le Monnier, A.; Zahar, J.-R. Gut microbiota, antibiotic therapy and antimicrobial resistance: A narrative review. Microorganisms 2020, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Limaye, P.B.; Renaud, H.J.; Klaassen, C.D. Effect of various antibiotics on modulation of intestinal microbiota and bile acid profile in mice. Toxicol. Appl. Pharmacol. 2014, 277, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of diet on the gut microbiota: Rethinking intervention duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [Green Version]
- Shadnoush, M.; Hosseini, R.S.; Khalilnezhad, A.; Navai, L.; Goudarzi, H.; Vaezjalali, M. Effects of probiotics on gut microbiota in patients with inflammatory bowel disease: A double-blind, placebo-controlled clinical trial. Korean J. Gastroenterol. 2015, 65, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- González, S.; Fernández-Navarro, T.; Arboleya, S.; de Los Reyes-Gavilán, C.; Salazar, N.; Gueimonde, M. Fermented dairy foods: Impact on intestinal microbiota and health-linked biomarkers. Front. Microbiol. 2019, 10, 1046. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Khatri, I.; Akhtar, A.; Subramanian, S.; Ramya, T. Metagenomics analysis reveals features unique to Indian distal gut microbiota. PLoS ONE 2020, 15, e0231197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Covington, A.; Pamer, E.G. The intestinal microbiota: Antibiotics, colonization resistance, and enteric pathogens. Immunol. Rev. 2017, 279, 90–105. [Google Scholar] [CrossRef]
- Wong, W.F.; Santiago, M. Microbial approaches for targeting antibiotic-resistant bacteria. Microb. Biotechnol. 2017, 10, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- Moghadam, M.T.; Amirmozafari, N.; Shariati, A.; Hallajzadeh, M.; Mirkalantari, S.; Khoshbayan, A.; Jazi, F.M. How phages overcome the challenges of drug resistant bacteria in clinical infections. Infect. Drug Resist. 2020, 13, 45. [Google Scholar] [CrossRef] [Green Version]
- Regeimbal, J.M.; Jacobs, A.C.; Corey, B.W.; Henry, M.S.; Thompson, M.G.; Pavlicek, R.L.; Quinones, J.; Hannah, R.M.; Ghebremedhin, M.; Crane, N.J. Personalized therapeutic cocktail of wild environmental phages rescues mice from Acinetobacter baumannii wound infections. Antimicrob. Agents Chemother. 2016, 60, 5806–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCune, V.; Quraishi, M.; Manzoor, S.; Moran, C.; Banavathi, K.; Steed, H.; Massey, D.; Trafford, G.; Iqbal, T.; Hawkey, P. Results from the first English stool bank using faecal microbiota transplant as a medicinal product for the treatment of Clostridioides difficile infection. EClinicalMedicine 2020, 20, 100301. [Google Scholar] [CrossRef] [PubMed]
- Terveer, E.M.; Van Beurden, Y.; Goorhuis, A.; Seegers, J.; Bauer, M.; Van Nood, E.; Dijkgraaf, M.; Mulder, C.; Vandenbroucke-Grauls, C.; Verspaget, H. How to: Establish and run a stool bank. Clin. Microbiol. Infect. 2017, 23, 924–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Taxonomic Rank | Control Group (%) | Diseased Group (%) | SD± | |
|---|---|---|---|---|
| Phylum | Bacteroidetes | 90.8 | 58.3 | 23 |
| Firmicutes | 9 | 11.2 | 1.6 | |
| Proteobacteria | 0.2 | 15.8 | 11.1 | |
| Actinobacteria | NA | 14.7 | NA | |
| Family | Bacteroidaceae | 0.4 | 18.7 | 12.9 |
| Prevotellaceae | 90.3 | 39.6 | 35.8 | |
| Lachnospiraceae | 5.7 | 0.4 | 3.7 | |
| Ruminococcaceae | 0.5 | 3.8 | 2.3 | |
| Veillonellaceae | 1.9 | NA | NA | |
| Enterobacteriaceae | 0.2 | 15.8 | 11.1 | |
| Bifidobacteriaceae | NA | 14.7 | NA | |
| Enterococcaceae | NA | 6.3 | NA | |
| Genus | Bacteroides | 0.4 | 18.7 | 12.9 |
| Prevotella | 90.3 | 39.6 | 35.8 | |
| Butyrivibrio | 2.1 | NA | NA | |
| Lachnospiraceae unclassified | 1.1 | NA | NA | |
| Roseburia | 2.2 | NA | NA | |
| Faecalibacterium | 0.5 | 3.8 | 2.4 | |
| Dialister | 1.8 | NA | NA | |
| Escherichia | 0.1 | 9.6 | 6.7 | |
| Klebsiella | 0.05 | 4.9 | 3.5 | |
| Bifidobacterium | NA | 14.7 | NA | |
| Enterococcus | NA | 6.3 | NA | |
| Species | Bacteroides vulgatus | 0.1 | 2.9 | 1.9 |
| Prevotella copri | 74.6 | 34.5 | 28.4 | |
| Prevotella sp. 885 | 1.7 | 0.9 | 0.6 | |
| Prevotella sp. AM42 24 | 8.2 | NA | NA | |
| Prevotella sp. CAG 279 | 1.3 | NA | NA | |
| Prevotella sp. CAG 520 | 3.6 | NA | NA | |
| Prevotella sp. CAG 5226 | 0.7 | 1.6 | 0.6 | |
| Prevotella stercorea | 0.2 | 2.6 | 1.8 | |
| Butyrivibrio crossotus | 2.1 | NA | NA | |
| Eubacterium rectale | 1.1 | NA | NA | |
| Roseburia faecis | 1.3 | NA | NA | |
| Faecalibacterium prausnitzii | 0.53 | 3.8 | 2.4 | |
| Dialister sp. CAG 357 | 1.8 | NA | NA | |
| E. coli | 0.1 | 9.6 | 6.7 | |
| Klebsiella pneumoniae | 0.05 | 3.1 | 2.1 | |
| Bifidobacterium longum | NA | 14.7 | NA | |
| Bacteroides dorei | NA | 13.3 | NA | |
| Bacteroides plebeius | NA | 1.2 | NA | |
| Enterococcus faecium | NA | 6.3 | NA | |
| Klebsiella quasipneumoniae | NA | 1.5 | NA |
| ARG Type | Resistome of Healthy Subjects | Resistome of Diseased Subjects |
|---|---|---|
| Tetracycline | tet32, tet40, tetA, tetO, tetQ, tetR, tetW | tetM, tetO, tetQ, tetS, tetW, tetA, tetB, tetR |
| Beta-lactam | aci1, blacfxA3, blacfxA6, blaCTX, blaTEM | blaCTX-M, blaCMH-1, blaCMY, blacfxA3, blacfxA6, blaNDM-1, blaOXY-1, blaTEM-1 |
| MLS 1 | MphA, ermF, ermB | ermB, ermF, ermX, mefA, MsrD |
| Sulphonamide | sul1, sul3 | sul1, sul2 |
| Aminoglycoside | aadA5, aph(3”)-Ib, aacA4 | aac6, aadA5, acrE, acrF, aph3 |
| MDR 2 | mdtA, emrK, mdtL | mdtG, mdtH, mdtN, mdtF, mdtC, mdtO, msbA, adeC, emrK, emrR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afridi, O.K.; Ali, J.; Chang, J.H. Fecal Microbiome and Resistome Profiling of Healthy and Diseased Pakistani Individuals Using Next-Generation Sequencing. Microorganisms 2021, 9, 616. https://doi.org/10.3390/microorganisms9030616
Afridi OK, Ali J, Chang JH. Fecal Microbiome and Resistome Profiling of Healthy and Diseased Pakistani Individuals Using Next-Generation Sequencing. Microorganisms. 2021; 9(3):616. https://doi.org/10.3390/microorganisms9030616
Chicago/Turabian StyleAfridi, Ome Kalsoom, Johar Ali, and Jeong Ho Chang. 2021. "Fecal Microbiome and Resistome Profiling of Healthy and Diseased Pakistani Individuals Using Next-Generation Sequencing" Microorganisms 9, no. 3: 616. https://doi.org/10.3390/microorganisms9030616
APA StyleAfridi, O. K., Ali, J., & Chang, J. H. (2021). Fecal Microbiome and Resistome Profiling of Healthy and Diseased Pakistani Individuals Using Next-Generation Sequencing. Microorganisms, 9(3), 616. https://doi.org/10.3390/microorganisms9030616